In this issue of Blood, 1 show that AML1-ETO (AE)-driven acute myeloid leukemia (AML) depends on phospholipase C-γ-1 (PLCG1). Although transcriptional repression of AML1-regulated genes is presumably the primary mechanism used by AE during leukemogenesis, this study provides evidence that AE directly induces the expression of PLCG1 and is a credible therapeutic target in this subset of AML.
The chromosomal translocation (8;21) is one of the most frequent genetic lesions in AML.2 The rearrangement results in expression of the AE fusion transcription factor, in which the DNA-binding domain of AML1 is fused with the transcriptional repressor ETO. Consequently, AE suppresses transcriptional activity of normal AML1 function, which drives myeloid differentiation and cell fate decisions. Therefore, its expression blocks differentiation and promotes self-renewal.3 However, expression of AE alone is insufficient for leukemogenesis in the absence of secondary cooperative mutations in genes, such as RAS, c-KIT, FLT3, and JAK2. Despite significant advances in understanding the molecular mechanism of the disease, conventional chemotherapy remains the mainstay treatment option for this group of AML, and most patients show favorable response rates and duration compared with other AML types. However, conventional chemotherapy is not curative, and disease relapse is common in a substantial number of patients.3
Previous studies using genome-wide analysis of transcription factor–binding sites and expression profiling upon genetic perturbation of AE or other transcription factors have, nonetheless, provided greater insights for the underlying mechanism driving leukemogenesis but thus far have not identified novel treatment options.3,4 To identify a druggable target, Schnoeder et al performed a global proteomic analysis using primary mouse leukemic stem cells expressing either AE or MLL-AF9. They noted altered expression of ∼868 genes. Subsequent in silico analysis revealed significant enrichment of calcium-dependent cellular signaling pathway with accompanied induction of PLCG1 expression only in AE-expressing leukemic cells. Further analysis using primary patient samples confirmed the elevated expression of PLCG1 exclusively in AE-positive AML. Importantly, high PLCG1 expression strongly correlated with poor therapeutic response and disease relapse, thus corroborating its role in leukemogenesis and disease relapse. Likewise, a previous study reported higher PLCG1 enzymatic activity in AE-expressing leukemic cells.5 Using human and mouse in vivo and in vitro models combined with primary human leukemia and normal hematopoietic cells, Schnoeder et al clearly demonstrate that PLCG1 is an indispensable mediator of AE-induced leukemogenesis. Perhaps more interestingly, genetic deletion of PLCG1 in mice did not have an adverse effect on normal hematopoiesis. Thus, it represents a relevant druggable target.
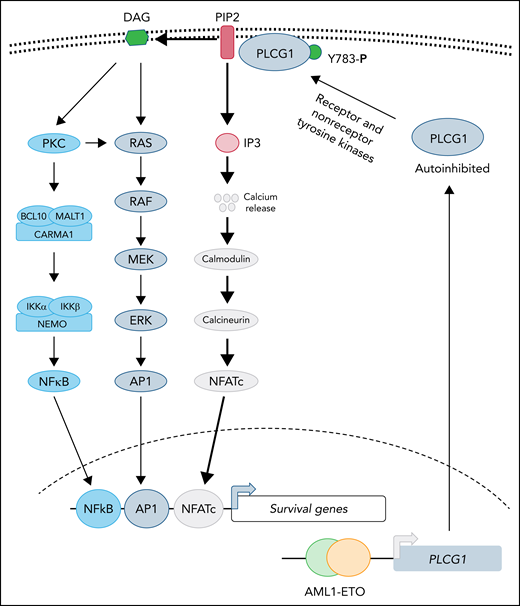
Recent structural studies demonstrate that PLCG1 is maintained in an autoinhibited state, and phosphorylation of tyrosine 783 stimulates phospholipase activity by relieving the autoinhibited conformation.6 Both receptor (epidermal growth factor receptor, B-cell receptor, and the linker for activation of T cells) and nonreceptor (Src, Syk, and Tec) tyrosine kinases have been reported to activate PLCG1 by directly phosphorylating tyrosine 783.6 Activated PLCG1 hydrolyzes membrane-bound phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) to generate the second messengers, diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), where DAG is retained in the membrane while IP3 diffuses and activates its receptors in the endoplasmic reticulum, leading to elevated cytosolic calcium that activates downstream signaling (see figure). Activation of PLCG1 has been implicated in many diseases. For instance, activated mutations in PCLG1 are frequently observed in both T- and B-cell leukemias, in which enhanced phospholipase activity promotes leukemogenesis by activating NFATc- and NF-κB–dependent transcription.7,8 However, in most cases, validation of elevated lipase activity of PLCG1 mutants has not been directly demonstrated. It is not clear whether overexpression of PLCG1 will be able to substitute for AE during leukemogenesis. The current study clearly demonstrates that depletion of PLCG1 suppresses the AE leukemic progression, which normally activates the NF-κB and MAPK pathways as well (see figure). It is not clear if elevated PLCG1 activates only calmodulin-calcineurin signaling in AE induced leukemia and how NF-κB and MAPK-AP1 networks cooperate during leukemogenesis with PLCG1 activation and in its absence.
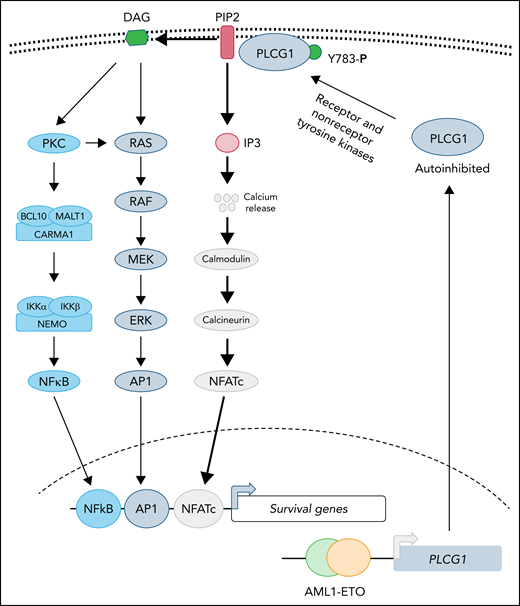
PLCG1 confers dependence in AE-driven leukemia. Expression of the AE fusion protein leads to upregulation of PLCG1, which is normally maintained in autoinhibited state. Activation of receptor or nonreceptor tyrosine kinases phosphorylates tyrosine 783 of PLCG1 activates its enzymatic activity. Activated PLCG1 hydrolyzes PIP2 to membrane-bound DAG and cytosolic IP3. Elevated IP3 activates calcium channel in the endoplasmic reticulum, leading to a cytosolic surge of calcium, which causes activation of calmodulin-calcineurin and NFATc activation. In parallel, DAG-mediated signaling leads to activation of NF-κB and AP1 transcription factor. Future studies will determine whether activation of NFATc is sufficient for AE-induced leukemogenesis and to what extent AP1 and NF-κB function is needed to support disease progression.
PLCG1 confers dependence in AE-driven leukemia. Expression of the AE fusion protein leads to upregulation of PLCG1, which is normally maintained in autoinhibited state. Activation of receptor or nonreceptor tyrosine kinases phosphorylates tyrosine 783 of PLCG1 activates its enzymatic activity. Activated PLCG1 hydrolyzes PIP2 to membrane-bound DAG and cytosolic IP3. Elevated IP3 activates calcium channel in the endoplasmic reticulum, leading to a cytosolic surge of calcium, which causes activation of calmodulin-calcineurin and NFATc activation. In parallel, DAG-mediated signaling leads to activation of NF-κB and AP1 transcription factor. Future studies will determine whether activation of NFATc is sufficient for AE-induced leukemogenesis and to what extent AP1 and NF-κB function is needed to support disease progression.
To gain further mechanistic insight, proteomic analysis using PLCG1-deficient cells underscored calcineurin-calcium signaling in AE cells. As proof of concept, treatment with cyclosporin A (CsA; a clinically approved calcineurin inhibitor that blocks intracellular calcium release) specifically halted leukemic progression with significant reduction in leukemic stem cells in AE-driven leukemia. As expected, MA9-driven leukemia did not show any response to CsA treatment, highlighting the specificity of this pathway to AE. Although CsA treatment significantly reduces the leukemic burden, it is not curative. Perhaps targeting additional druggable nodes in combination, such as BCL29 and AP1,10 may provide a curative response.
Nevertheless, several key questions remain. (1) How is PLCG1 activated? Is its overexpression sufficient to relieve the autoinhibited state or does it still depend on tyrosine kinase–mediated activation? (2) How does PLCG1 regulate transcriptional activity during AE-induced leukemogenesis? (3) How do NF-κB, AP1, and NFATc regulate transcriptional machinery under PLCG1 depletion? Perhaps a global phosphoproteomic and Chip-seq for AP1, NFATc, and NF-κB in PLCG1 knockout cells may provide deeper insights into the underlying mechanism, which may help develop better therapeutic strategy. Given the paucity in development of effective treatments for AML, the current study provides a strong rationale for evaluating CsA in AE-positive AML with conventional chemotherapy that may induce more durable responses.
Conflict-of-interest disclosure: The author declares no competing financial interests.