Abstract
We have expressed a soluble N-glycosylated form of the murine interleukin-11 (IL-11) receptor α-chain (sIL-11R) and examined signaling in cells expressing the gp130 molecule. In the presence of gp130 but not the transmembrane IL-11R, the sIL-11R mediated IL-11–dependent differentiation of M1 leukemic cells and proliferation in Ba/F3 cells. Early intracellular events stimulated by the sIL-11R including phosphorylation of gp130, STAT 3, and SHP-2 were similar to signaling through the transmembrane IL-11R. IL-11 bound to sIL-11R with low affinity (kd 10 to 50 nmol/L). Binding of sIL-11R to gp130 was IL-11 dependent with intermediate affinity (kd 1.5 to 3.0 nmol/L). However, the concentration of IL-11 required for signaling through the sIL-11R was 10- to 20-fold greater than that required for cells expressing the transmembrane IL-11R and gp130 in the absence of sIL-11R. Furthermore, the sIL-11R was capable of antagonizing the activity of IL-11 when tested on cells expressing the transmembrane IL-11R and gp130. We propose that the observed IL-11 antagonism by the sIL-11R may depend on limiting numbers of gp130 molecules on cells already expressing the transmembrane IL-11R.
INTERLEUKIN-11 (IL-11) was first identified because of its ability to support the proliferation of an IL-6–dependent plasma cell line.1 Its biological actions include the ability to stimulate proliferation of multipotential hemopoietic progenitor cells,2 the enhancement of megakaryocyte and platelet formation,3-5 and the stimulation of acute phase protein synthesis.6 Many of these actions are shared with IL-6, leukemia inhibitory factor (LIF ), oncostatin M (OSM), cardiotrophin-1 (CT-1), and ciliary neurotrophic factor (CNTF ).7-11 The overlapping functions of these cytokines is in part due to the sharing of the cell surface receptor component, gp1309 with specificity being determined by a specific low-affinity receptor α-chain.12
The IL-11 receptor α-chain (IL-11R) has recently been cloned and shown to share many structural and functional similarities with the IL-6 receptor α-chain (IL-6R).13 Firstly, the extracellular domain shows 24% amino acid identity including the characteristic conserved Trp-Ser-X-Trp-Ser (WSXWS) motif. The short cytoplasmic domain (34 amino acids) lacks the Box 1 and 2 regions that are required for activation of the JAK/STAT signaling pathway. Secondly, the IL-11R binds its ligand with a low affinity (kd ∼10 nmol/L) and alone is insufficient to transduce a biological signal. The generation of a high affinity receptor (kd ∼400 to 800 pmol/L) capable of signal transduction requires coexpression of the IL-11R and gp130.13,14 In contrast, the high-affinity receptors for LIF, CT-1, and CNTF signal through a heterodimer of the LIF receptor and gp130.10,15-17 Finally, the early signaling pathways of IL-6 and IL-11 appear similar and include phosphorylation of the JAKs, STAT1 and 3, and activation of the mitogen-activated protein (MAP) kinase pathway.18 19
Naturally occurring soluble receptors, consisting of the extracellular domain, have been described for many cytokine receptors including the specific low-affinity receptor α-chains for IL-6 and LIF.20,21 The soluble forms bind their respective ligand with a similar low affinity (1 to 20 nmol/L) to their transmembrane α-chain counterparts in the absence of gp130.20,22,23 Soluble IL-6R (sIL-6R) can associate with cell surface gp130 in the presence of IL-6 and transduce a signal.23 A soluble form of the mouse LIFR is generated by alternative splicing of mRNA transcripts.24 In contrast to the sIL-6R, soluble LIFR can bind LIF but is unable to transduce a signal and thus behaves as a LIF antagonist.20 Recently, a transcript potentially encoding a soluble IL-11R (sIL-11R) has been identified25 although the naturally occurring protein has yet to be isolated. Recombinant forms of the sIL-11R, in the presence of IL-11, have been shown to mediate an IL-11–type signal in cells that express gp130, but not the transmembrane IL-11R. In cells expressing the transmembrane IL-11R, the sIL-11R augments the IL-11 response.26-28 These results suggest that IL-11R is very similar to the IL-6R. However, in vitro studies using tagged recombinant forms of IL-11, soluble human IL-11 receptor, and gp130 have suggested that dimers of the IL-11/sIL-11R complex bind to a single gp130 molecule rather than two like that seen with the IL-6–signaling complex.28
The experiments reported here were undertaken to examine the action of a recombinant sIL-11R expressed in the Chinese hamster ovary (CHO) cell line. Confirming recent reports, we show that the sIL-11R, in the presence of IL-11 mediated a proliferative or differentiative signal in appropriate cell lines expressing gp130 alone. However, the sIL-11R was capable of antagonizing the IL-11 response of cells expressing gp130 and the transmembrane IL-11R. The antagonistic activity may have implications for the predicted in vivo effects of sIL-11R.
MATERIALS AND METHODS
Construction of a soluble murine IL-11R cDNA. The extracellular domain of the murine IL-11R was amplified using Taq polymerase from a full length, previously described cDNA clone.13 The 5′ and 3′ ends of the cDNA were modified to include Xba I sites. The predicted protein began at amino acid residue Ser-24 in the sequence described by Hilton et al.13 A stop codon was inserted immediately 5′ of the transmembrane domain after Gln-367. The cDNA was then cloned into the Xba I site of a modified pEFBOS vector containing the IL-3–signal sequence followed by an inframe FLAG sequence (DYKDDDDK) so that the N-terminus of the predicted secreted protein was DYKDDDDKSPCPQA. The nucleotide sequence of the plasmid pEFBOS/sIL-11R was confirmed by dideoxy sequencing using a PRISM (Applied Biosystems, Inc, Foster City, CA) Ready Reaction DyeDeoxy Terminator Cycle Sequencing kit on an Applied Biosystems 373 DNA sequencer (Foster City, CA).
Expression and purification of sIL-11R. CHO cells (4 × 106) were washed twice in ice cold phosphate-buffered saline (PBS) and resuspended in PBS at 5 × 106 cells/mL. Cells were aliquoted into a 0.4-cm electroporation cuvette with 20 μg of pEFBOS/sIL-11R and 2 μg pPGKneo and then electroporated at 270 V and 960 mF in a Bio-Rad Gene-Pulser (Bio-Rad Laboratories, Hercules, CA). Cells were centrifuged through 1 mL fetal calf serum (FCS), resuspended in RPMI-1640 medium containing 5% (vol/vol) FCS, and plated onto 10-cm diameter petri dishes. After 48 hours, geneticin (1 mg/mL) was added and 10 to 14 days later, resistant clones were picked. The highest sIL-11R expressing clone was identified by Western blot analysis.
Conditioned medium was concentrated 10× using a YM-10 membrane (Amicon, Beverly, MA) and applied to an M2 anti-FLAG antibody affinity column (Eastman Kodak, New Haven, CT). After washing with 10 mmol/L Tris in PBS (pH 8.0) containing 0.02% (wt/vol) Tween-20 and 0.02% (wt/vol) sodium azide, bound sIL-11R was eluted from the column with 8 mL of 62.5 μg/mL FLAG peptide (Eastman Kodak). sIL-11R containing fractions were determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting with M2 anti-FLAG antibody and exchanged into 150 mmol/L NaCl containing 0.02% Tween 20 using gel filtration PD-10 columns (Pharmacia, Uppsala, Sweden). Fractions containing immunoreactive material were pooled and purified by gel filtration using a Superdex 75 column (Pharmacia). Protein estimation was performed by measuring the optical density (590 nm) using Coomassie stain (Pierce, Rockford, IL) and compared with a bovine serum albumin standard. N- terminal sequencing analysis was performed using a Beckman 6300 high performance amino acid analyzer (Irvine, CA) as previously described.29
Immunoprecipitations. For antiphosphotyrosine assays, cell lysates from 1 × 107 cells were prepared using 1% Triton X-100 (Sigma, St Louis, MO), 150 mmol/L NaCl, 50 mmol/L Tris HCl, 2 mmol/L EDTA, 1 mmol/L sodium vanadate, 2 μg/mL aprotinin, and 100 μg/mL phenylmethylsulfonyl fluoride. Five microliters of the antiphosphotyrosine antibody (4G10, UBI) or anti-LIF receptor antibody (Regeneron Pharmaceuticals, Tarrytown, NY) was added and the antibody immobilized using Protein A sepharose. After incubation at 4°C overnight, pellets were washed with 1 mL of lysis buffer containing 1% Nonidet P-40 (BDH Laboratory Supplies, Poole, UK), 2 μg/mL aprotinin, and 1 mmol/L sodium vanadate and then resuspended in 30 μL of reducing or nonreducing SDS sample buffer.
Western blotting. Samples were run on 10% SDS-PAGE and electrophoretically transferred onto prewetted polyvinylidene difluoride (PVDF )-plus (Micron Separations Inc, Westborough, MA). Membranes were blocked with 5% (wt/vol) skim milk in PBS. Phosphotyrosine containing proteins were detected by incubation with gp130 (UBI), STAT3 (Santa Cruz Biotechnology, Inc, Santa Cruz, CA) or SHP-2 (Transduction Laboratories, Lexington, Kentucky) antibodies and bound antibodies detected using goat antirabbit IgG conjugated to horseradish peroxidase. FLAG containing proteins were detected by incubation with the mouse M2 anti-FLAG antibody and then horseradish peroxidase conjugated rabbit anti-mouse antibody (DAKO, Glostrup, Denmark). Bound antibody was visualized using the ECL substrate kit (Amersham, Buckinghamshire, UK) followed by autoradiography.
PAGE. Electrophoreses were performed using Bio-Rad 8% to 25% SDS-PAGE precast Pharmacia Phast-Gels according to the manufacturer's instructions and the gels were visualized by silver-staining.
Deglycosylation. Four micrograms of lyophilized sIL-11R was deglycosylated using the enzymatic deglycosylation kit (GLYKO, Movato, CA). Samples were treated for 1 hour with O-glycosidase DS and NANaseII (GLYKO) and followed by N-glycanase F for 3 hours at 37°C. One microliter of reaction samples were then run on 8% to 25% SDS-PAGE Phast-Gel under nonreducing conditions.
Binding assays. One to two micrograms sIL-11R was radio-iodinated using a modified iodine monochloride method as previously described.30 Specific radioactivity was estimated to be 25,000 counts per minute (cpm)/ng. 2 × 107 cells/mL in RPMI-1640 medium containing 10% (vol/vol) FCS and 20 mmo/L HEPES were incubated at room temperature for 4 hours with various combinations of 125I-sIL-11R and hIL-11 in the presence or absence of cold competitor sIL-11R (100-fold excess). Cell associated and free 125I-sIL-11R were separated by rapid centrifugation through 200 μL of FCS and quantitated in a γ-counter. For estimation of the affinity of IL-11 for the sIL-11R, 2 mg/mL of sIL-11R was immobilized using anti-FLAG coated beads and a titration of unlabeled IL-11 performed in the presence of 125I-labeled IL-11 (2 × 106 cpm/mL). The bindability of the 125I-labeled IL-11 was only 10%. This was accounted for when estimation of affinities by the nonlinear curve fitting program (LIGAND) was performed.
Cytokines and biological assays. Human IL-11 and oncostatin M were purchased from R & D Systems (Minneapolis, MN). Cytospins of M1 cells after 5 days in liquid culture were performed as previously described.14,313H-Thymidine assays were performed by adding M1 or M1/IL-11R cells (1 × 104 /mL) into 96-well plates and then adding cytokines as indicated. Plates were incubated in humidified air with 10% CO2 at 37°C. 3H-Thymidine (1 to 2 μCi/well) was added after 4 days and 16 hours later, cells were procured and cell associated radioactivity measured. The proliferation of Ba/F3 cells in response to cytokines was measured in LUX 60 microwell HL-A plate (Nunc Inc, Naperville, IL) and semisolid agar assays to measure differentiation of M1 cells were performed as described.13
RESULTS
Production and purification of sIL-11R. A stable CHO cell line producing sIL-11R was established by coelectroporation of the sense sIL-11R cDNA with pPGK neo. The supernatant was purified using an anti-FLAG affinity column. Western blots of the FLAG-eluted CHO cell fractions showed immunoreactive material of approximately 50 kD and 200 kD. When fractions were Western blotted from gels run under reducing conditions, the 200-kD band was not seen (Fig 1A). This suggested that the 200-kD band represented disulphide-linked multimers of the sIL-11R. Purification of pooled fractions 4 to 6 by gel filtration revealed a peak at 44 kD (Fig 1B). Silver staining of an SDS-PAGE revealed at least 4 distinct bands of between 40 and 55 kD (Fig 1C). Although the primary amino acid sequence predicts a protein of 30 kD, the extracellular domain of the sIL-11R contains 2 potential N-glycosylation sites. To determine whether these multiple bands were due either to differential glycosylation or contamination, the pooled sample was lyophilized and then treated with deglycosylation enzymes. Treatment with N-glycanase F, which removes N-linked carbohydrates, reduced all bands to a single 36-kD species (Fig 1D, lane 2). This suggested the presence of multiple N-linked glycosylation species. The preparation was also analyzed by N-terminal sequencing, which demonstrated the presence of the expected 24 N terminal amino acids (DYKDDDESRSPXPQAWGPPGVQYG). Thus, silver stained gels and amino acid sequencing confirmed >95% purity of the sIL-11R sample subsequently used for biological and biochemical experiments.

Purification of sIL-11R. (A) Western blot of CHO sIL-11R eluates from anti-FLAG M2 affinity column. Numbers above represent fractions (0.5 mL) eluted with FLAG peptide (50 μg/mL). SM, starting material; BT, breakthrough when loaded onto affinity column. Blots probed with anti-FLAG antibody. Samples prepared by mixing with equal volume of 2× nonreducing (left side of gel) or reducing (right side of gel) sample buffer and run on 8% to 25% SDS-PAGE. Molecular weights on left side (kD). (B) Chromatogram of Superdex 75 gel filtration (Pharmacia) of pooled fractions 4 to 6. (C) Silver stain of nonreducing SDS-PAGE of fractions 40 through 44 from gel filtration. (D) Silver stain gel of pooled fractions before (lane 1) and after (lane 2) deglycosylation with N-glycanase F as described in experimental methods.
Purification of sIL-11R. (A) Western blot of CHO sIL-11R eluates from anti-FLAG M2 affinity column. Numbers above represent fractions (0.5 mL) eluted with FLAG peptide (50 μg/mL). SM, starting material; BT, breakthrough when loaded onto affinity column. Blots probed with anti-FLAG antibody. Samples prepared by mixing with equal volume of 2× nonreducing (left side of gel) or reducing (right side of gel) sample buffer and run on 8% to 25% SDS-PAGE. Molecular weights on left side (kD). (B) Chromatogram of Superdex 75 gel filtration (Pharmacia) of pooled fractions 4 to 6. (C) Silver stain of nonreducing SDS-PAGE of fractions 40 through 44 from gel filtration. (D) Silver stain gel of pooled fractions before (lane 1) and after (lane 2) deglycosylation with N-glycanase F as described in experimental methods.
sIL-11R signaling is comparable to that of the transmembrane IL-11R. The biological activity of the sIL-11R was examined using the murine myeloid cell line, M1, which expresses gp130 and the α-chain receptors for LIF and IL-6 but not for IL-11. After 4 days in suspension culture, 79 ± 25% of cells stimulated with IL-11 and sIL-11R showed morphological features of macrophage differentiation (mean ± SD of 3 experiments, Fig 2D). This change was associated with inhibition of cellular proliferation as evidenced by reduced incorporation of 3H-thymidine (see below). In contrast, cells stimulated with IL-11 or sIL-11R alone showed no significant differentiation (Fig 2A and B). As reported previously,13 the majority of cells (93 ± 4%) stimulated with LIF showed a macrophage phenotype (Fig 2C) as did cells that expressed the transmembrane IL-11R when treated with IL-11 (92 ± 5%, data not shown). Flourescence-activated cell sorter (FACS) analysis of these cells confirmed increased surface expression of Mac-1α to levels similar to those observed with LIF treatment (data not shown).

sIL-11R induces differentiation of M1 cells in the presence of IL-11. May-Grünwald Giemsa Stain (×400 original magnification) of parental M1 cells after 5 days in liquid culture in the presence of (A) IL-11 500 pmol/L (B) sIL-11R 40 nmol/L (C) LIF 1,000 U/mL (D) IL-11 plus sIL-11R.
sIL-11R induces differentiation of M1 cells in the presence of IL-11. May-Grünwald Giemsa Stain (×400 original magnification) of parental M1 cells after 5 days in liquid culture in the presence of (A) IL-11 500 pmol/L (B) sIL-11R 40 nmol/L (C) LIF 1,000 U/mL (D) IL-11 plus sIL-11R.
Experiments were then performed to examine the early phosphorylation events induced by sIL-11R. Lysates from M1 cells stimulated with various cytokines were immunoprecipitated with either an anti-LIF receptor or an antiphosphotyrosine antibody. Immunoprecipitates were run on SDS-PAGE gels, Western blotted, and probed with either the antiphosphotyrosine antibody in the case of the anti-LIF immunoprecipitate (Fig 3A) or specific anti-gp130, STAT 3, or SHP-2 antibodies in the case of the antiphosphotyrosine immunopricipitates (Fig 3B through D). As expected, stimulation with maximal doses LIF (1,000 U/mL) or OSM(10 ng/mL) resulted in phosphorylation of a 210-kD protein, which was confirmed to be the LIF receptor α-chain by immunoprecipitation (Fig 3A). Like IL-6, IL-11 (5 nmol/L) in the presence of sIL-11R (40 nmol/L) did not result in phosphorylation of the LIF receptor α-chain (Fig 3A). However, consistent with the biological effects above, cells stimulated with sIL-11R and IL-11 showed phosphorylation of gp130 and STAT 3 (Fig 3B and C). Probing for the tyrosine phosphatase SHP-2 also showed phosphorylation in response to sIL-11R in the presence of IL-11 (Fig 3D). The phosphorylation pattern in cells stimulated with IL-11 or sIL-11R alone was no different to saline controls. These experiments suggested that IL-11 plus sIL-11R induced a differentiative response in M1 cells via phosphorylation of gp130. Thus, analogous to the transmembrane IL-11R and similar to results with IL-6 and the sIL-6R,23 the sIL-11R acted as an agonist to effectively transmit an IL-11 signal. This showed that the intracellular domain of IL-11R was not required for a biological response.

sIL-11R–stimulated tyrosine phosphorylation of gp130, STAT 3, and SHP-2. Western blot of cell lysates following immunoprecipitation with either (A) anti-LIF receptor antibody or (B through D) antiphosphotyrosine antibody. Blots were then probed with (A) antiphosphotyrosine, (B) anti-gp130, (C) anti-STAT 3, or (D) anti-SHP-2 antibodies. Lysates are from parental M1 cells in the presence of saline, LIF (1,000 U/mL), IL-6 (100 ng/mL), oncostatin M (100 ng/mL), IL-11 (5 nmol/L), sIL-11R plus IL-11 or sIL-11R alone (40 nmol/L). Samples were run on 8% to 25% SDS-PAGE.
sIL-11R–stimulated tyrosine phosphorylation of gp130, STAT 3, and SHP-2. Western blot of cell lysates following immunoprecipitation with either (A) anti-LIF receptor antibody or (B through D) antiphosphotyrosine antibody. Blots were then probed with (A) antiphosphotyrosine, (B) anti-gp130, (C) anti-STAT 3, or (D) anti-SHP-2 antibodies. Lysates are from parental M1 cells in the presence of saline, LIF (1,000 U/mL), IL-6 (100 ng/mL), oncostatin M (100 ng/mL), IL-11 (5 nmol/L), sIL-11R plus IL-11 or sIL-11R alone (40 nmol/L). Samples were run on 8% to 25% SDS-PAGE.
Signaling by sIL-11R requires higher concentrations of IL-11 than by the transmembrane IL-11R. To quantitate the biological activity of the sIL-11R, we examined its ability to induce differentiation to postmitotic macrophages and thereby inhibit thymidine incorporation of M1 cells.31 32 Cells were stimulated for 5 days with sIL-11R plus IL-11. Titration of sIL-11R in the presence of IL-11 (500 pmol/L) showed a maximal biological response in the presence of 40 nmol/L sIL-11R (Fig 4A). In the presence of 40 nmol/L sIL-11R, an IL-11 titration was performed and compared with the dose-response for M1 cells expressing the transmembrane IL-11R in addition to gp130 (M1/IL-11R). The sIL-11R produced a similar degree of inhibition of thymidine incorporation to that seen with IL-11 on M1/IL-11R cells (Fig 4B). However, the concentration of IL-11 required to produce a 50% maximal effect on M1 cells was 375 ± 150 pmol/L in the presence of the sIL-11R compared with 15 ± 10 pmol/L on M1/IL-11R cells in the absence of sIL-11R (mean ± SD of 3 experiments). This 10- to 20-fold difference in IL-11 sensitivity was not due to clonal variation of the M1 or the M1/IL-11R cells as the same dose response was observed with 3 independent clones (data not shown).

Biological activity of sIL-11R on M1 cells.(A) sIL-11R titration in the presence of saline (□) or 500 pmol/L IL-11 (▪). (B) IL-11 titration in the presence of 40 nmol/L sIL-11R (□) compared with cells expressing the transmembrane IL-11R (▪) in the absence of sIL-11R. Dose response measured by the ability to induce differentiation in M1 cells and thereby reduce 3H-thymidine incorporation. Results expressed as mean and standard deviation of triplicate wells from 2 representative experiments.
Biological activity of sIL-11R on M1 cells.(A) sIL-11R titration in the presence of saline (□) or 500 pmol/L IL-11 (▪). (B) IL-11 titration in the presence of 40 nmol/L sIL-11R (□) compared with cells expressing the transmembrane IL-11R (▪) in the absence of sIL-11R. Dose response measured by the ability to induce differentiation in M1 cells and thereby reduce 3H-thymidine incorporation. Results expressed as mean and standard deviation of triplicate wells from 2 representative experiments.
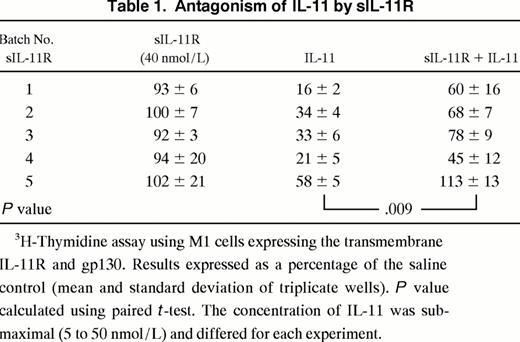
Effects of sIL-11R in the presence of transmembrane IL-11R. The activity of sIL-11R was then examined on M1/IL-11R cells. In view of the agonist activity of the sIL-11R on M1 cells we expected the presence of the sIL-11R would augment the IL-11 response of M1 cells expressing the transmembrane IL-11R. Surprisingly, sIL-11R reproducibly inhibited the action of IL-11 on M1/IL-11R cells. When sIL-11R (40 nmol/L) was added to IL-11, there was a consistent inhibition of IL-11 activity as evidenced by a shift in the dose-response curve (Fig 5A). Of note, the antagonism could be overcome by increasing the concentration of IL-11. However, at no IL-11 concentration did the sIL-11R augment the response of M1/IL-11R cells to IL-11. The IL-11 antagonism was reproducible using 5 independently purified batches of sIL-11R (Table 1). Specificity of the IL-11 antagonism was examined by combining sIL-11R with LIF. The sIL-11R had no effect on the ability of LIF to induce differentiation and thereby inhibit thymidine incorporation of M1/IL-11R cells (Fig 5B). This suggested that the inhibition by sIL-11R was specific for IL-11 and was not, for example, a result of the sIL-11R interacting with gp130 or nonspecific toxicity.

Biological activity of sIL-11R on M1/IL-11R cells. 3H-thymidine incorporation (1 μCi/well) measuring (A) IL-11 dose response in the presence of saline (□) or 40 nmol/L sIL-11R (▪) or (B) sIL-11R dose response in the presence of 20 pmol/L IL-11 (•) or 25 U/mL LIF (○). (C) Effect of sIL-11R on colony formation in agar cultures. Shaded region of bar represents the number of differentiated colonies. Three hundred cells added/plate. Results expressed as the mean and standard deviation of duplicate cultures from 2 independent experiments.
Biological activity of sIL-11R on M1/IL-11R cells. 3H-thymidine incorporation (1 μCi/well) measuring (A) IL-11 dose response in the presence of saline (□) or 40 nmol/L sIL-11R (▪) or (B) sIL-11R dose response in the presence of 20 pmol/L IL-11 (•) or 25 U/mL LIF (○). (C) Effect of sIL-11R on colony formation in agar cultures. Shaded region of bar represents the number of differentiated colonies. Three hundred cells added/plate. Results expressed as the mean and standard deviation of duplicate cultures from 2 independent experiments.
Agar culture experiments were also performed to determine whether the effects observed in thymidine proliferation assays were seen at the clonal level and where differentiation could be monitored by cell migration through agar. Consistent with the thymidine assays, the sIL-11R was able to partially antagonize the clonal suppression and differentiation induced by the IL-11 (Fig 5C). IL-11 (50 pmol/L) alone induced 100% differentiation of colonies while the combination of sIL-11R (40 nmol/L) and IL-11 (50 pmol/L) induced only 65% differentiated colonies. Colony size was also significantly larger compared with IL-11 alone (data not shown).
To further confirm that the sIL-11R inhibited an IL-11 response in cells expressing the transmembrane IL-11R and gp130, we examined the effect on early phosphorylation events in M1/IL-11R cells. Consistent with the thymidine uptake and agar experiments, there was a reduction in STAT 3 phosphorylation in M1/IL-11R cells treated with sIL-11R (40 nmol/L) and IL-11 (100 pmol/L) compared with IL-11 alone at the same concentration (data not shown).
The agonist and antagonist activities of sIL-11R are not cell-type specific. To show that the sIL-11R activities were not cell-type specific, we also examined the activities on the IL-3–dependent cell line, Ba/F3. A Ba/F3 cell line expressing gp130 was able to proliferate in response to sIL-11R only in the presence of IL-11 (Fig 6A) thus confirming the agonist activity seen on M1 cells. Consistent with the results in M1/IL-11R cells, sIL-11R inhibited the activity of IL-11 (Fig 6B) on Ba/F3 cells expressing IL-11R. Specificity of the antagonism was shown by the inability of sIL-11R to antagonize the IL-3 response. Unlike the M1/IL-11R cells, the IL-11 antagonism was not overcome by maximal concentrations of IL-11. These results confirmed that the sIL-11R could either mediate or inhibit not only a differentiative response (M1 cells) but also a proliferative signal in Ba/F3 cells (Fig 6).

Activity of sIL-11R on Ba/F3 cell proliferation. Microwell assay with 200 cells added per well. Cells were visually counted and results expressed as a percentage of the maximal response (≥200 cells at 48 hours). (A) Ba/F3 cells expressing gp130 stimulated with sIL-11R alone (○) or combined with 500 pmol/L IL-11 (•). (B) Ba/F3 cells expressing gp130 and the transmembrane IL-11R stimulated with IL-11 alone (□) or combined with 40 nmol/L sIL-11R (▪). Control cultures stimulated with IL-3 alone (○) or combined with 40 nmol/L sIL-11R (•) are also shown.
Activity of sIL-11R on Ba/F3 cell proliferation. Microwell assay with 200 cells added per well. Cells were visually counted and results expressed as a percentage of the maximal response (≥200 cells at 48 hours). (A) Ba/F3 cells expressing gp130 stimulated with sIL-11R alone (○) or combined with 500 pmol/L IL-11 (•). (B) Ba/F3 cells expressing gp130 and the transmembrane IL-11R stimulated with IL-11 alone (□) or combined with 40 nmol/L sIL-11R (▪). Control cultures stimulated with IL-3 alone (○) or combined with 40 nmol/L sIL-11R (•) are also shown.
Monomeric sIL-11R can antagonize IL-11 response. A possible explanation for the IL-11 antagonism was the presence of high molecular weight aggregates (Fig 1A) binding IL-11 but unable to bind to membrane bound gp130. To exclude this possibility, we separated the high molecular weight forms from the monomeric sIL-11R by gel filtration using a Superdex 75 Column (Pharmacia) (Fig 7A). We then compared the activity of both forms on M1/IL-11R cells in the absence or presence of 50% maximal concentration of IL-11. Compared with the high molecular weight forms, the monomeric form of sIL-11R was equally capable of inhibiting the activity of IL-11 on M1/IL-11R cells (Fig 7B). Thus, the IL-11 anagonism was not simply due to the presence of aggregated forms ‘mopping’ up IL-11 without binding to gp130.

IL-11 antagonism by monomeric and aggregated forms of sIL-11R. (A) Western blot of sIL-11R fractions following gel filtration by Superdex 75 column (Pharmacia). For the bioassay, fractions 16 and 17 were used for the high moelcular weight form and fractions 20 to 22 for the monomeric form. Molecular weight markers are shown on the left side. (B) 3H-thymidine incorporation (1 μCi/well) measuring the response of M1/IL-11R cells to sIL-11R in the absence (□) or presence (▪) of 10 pmol/L IL-11. The monomeric form of sIL-11R is the solid line and the high molecular weight form is the broken line.
IL-11 antagonism by monomeric and aggregated forms of sIL-11R. (A) Western blot of sIL-11R fractions following gel filtration by Superdex 75 column (Pharmacia). For the bioassay, fractions 16 and 17 were used for the high moelcular weight form and fractions 20 to 22 for the monomeric form. Molecular weight markers are shown on the left side. (B) 3H-thymidine incorporation (1 μCi/well) measuring the response of M1/IL-11R cells to sIL-11R in the absence (□) or presence (▪) of 10 pmol/L IL-11. The monomeric form of sIL-11R is the solid line and the high molecular weight form is the broken line.
Binding affinity of the sIL-11R. Receptor binding assays were performed to determine the affinity of IL-11 for the sIL-11R. Previous binding experiments have shown the affinity of IL-11 for Ba/F3 cells expressing the transmembrane IL-11R in the absence of gp130 is low (kd ∼10 nmol/L).13 When sIL-11R was immobilized on anti-FLAG labeled beads, the affinity of IL-11 for sIL-11R was similarly low (kd ∼10 to 50 nmol/L, data not shown).
The binding affinity of the IL-11/sIL-11R complex for membrane bound gp130 was estimated by binding of 125I-labeled sIL-11R to M1 cells. Initial experiments determined that a minimum IL-11 concentration of 5 to 10 nmol/L was required for detectable binding of 125I-labeled sIL-11R to M1 cells (Fig 8A). In the presence of a maximal concentration of IL-11 (50 nmol/L), the affinity of interaction of the IL-11/sIL-11R complex for gp130 was estimated by Scatchard analysis to be 1.5 to 3.0 nmol/L (Fig 8B, n = 3). Experiments using immobilized gp130 on a biosensor-chip showed binding of the IL-11/sIL-11R complex but not IL-11 or sIL-11R alone to the immobilized gp130. Assuming a kd of 50 nmol/L for IL-11/sIL-11R interaction, the calculated affinity of the IL-11/sIL-11R complex for gp130 was slightly less than that calculated using 125I-labeled sIL-11R. The kd was 10 nmol/L with a KA of 9.8 × 107 mol/L−1 (data not shown). These rates are similar to those observed with a monomeric IL-6/sIL-6R complex.33

Binding characteristics of sIL-11R. (A) Effect of IL-11 concentration on specific binding of 125I-labeled sIL-11R (2 × 105 cpm) to 106 M1 cells. (B) Scatchard analysis of sIL-11R binding to M1 cells in the presence of IL-11 (50 nmol/L). (C) Effect of 125I-labeled IL-11 (1.5 nmol/L) binding to 106 M1/IL-11R cells in the absence (□) or presence () of 30-fold excess of unlabeled IL-11. Results are mean and standard deviation of duplicate measurements.
Binding characteristics of sIL-11R. (A) Effect of IL-11 concentration on specific binding of 125I-labeled sIL-11R (2 × 105 cpm) to 106 M1 cells. (B) Scatchard analysis of sIL-11R binding to M1 cells in the presence of IL-11 (50 nmol/L). (C) Effect of 125I-labeled IL-11 (1.5 nmol/L) binding to 106 M1/IL-11R cells in the absence (□) or presence () of 30-fold excess of unlabeled IL-11. Results are mean and standard deviation of duplicate measurements.
To confirm that the anti-IL-11 activity of the sIL-11R was due to inhibition of IL-11 binding to the transmembrane IL-11R, we measured the binding of 125I-labeled IL-11 (150 pmol/L) to M1/IL-11R cells in the presence of untransfected CHO cell supernatant or sIL-11R (80 nmol/L). These conditions were used because we could not detect binding of sIL-11R on M1 cells at this concentration of IL-11 (Fig 8A). The sIL-11R but not untransfected CHO cell supernatant inhibited specific IL-11 binding on M1/IL-11R cells (Fig 8C). The nonspecific binding in these assays was high relative to the specific IL-11 binding because IL-11 bindability was only 10%. Nevertheless, this inhibition of binding was seen in 3 consecutive experiments. These results suggested that the sIL-11R could bind IL-11 but not gp130 at that concentration of IL-11 and thus compete with the transmembrane IL-11R for ligand. Given that the affinity of IL-11 for sIL-11R and transmembrane IL-11R was similar, competition for IL-11 would depend on the relative numbers of soluble and transmembrane receptors. The number of transmembrane IL-11R molecules per M1/IL-11R cell was estimated to be not more than 8,000.13 Thus, we estimated the number of soluble IL-11R molecules in this binding assay to be at least 10-fold greater than the number of transmembrane IL-11R molecules.
DISCUSSION
We have produced an affinity-purified soluble form of the murine IL-11 receptor α-chain in CHO cells. Silver stained SDS-PAGE and N-terminal amino acid sequencing suggest that the material was >95% pure (Fig 1). Analogous to IL-6/sIL-6R, the combination of IL-11 and sIL-11R mediated a proliferative (Ba/F3 cells) or differentiative (M1 cells) signal in cells expressing gp130 without the transmembrane IL-11R (Figs 2-4 and Fig 6). These data confirm recent reports that the cytoplasmic domain of the IL-11R is not required for IL-11 signaling.26-28
However, experiments using cell lines expressing gp130 and the transmembrane IL-11R yielded surprising results. Firstly, the concentration of IL-11 required to mediate a biological response via the sIL-11R was 10- to 20-fold greater than via the transmembrane receptor (Fig 4B). Despite this, the biological response using the sIL-11R was qualitatively and quantitatively similar to that seen with the transmembrane receptor. A similar requirement for an increased concentration of ligand has been noted previously for the sIL-6R.23 The observation that a cytoplasmic deletion mutant (transmembrane domain still present) of the IL-6R did not exhibit an increased IL-6 requirement suggests this phenomenon is not due to the lack of the cytoplasmic domain. Baumann et al26 also noted a similar requirement for an increased concentration of IL-11 to stimulate cells through the sIL-11R. They suggested that this may be due to submaximal concentrations of sIL-11R as they were unable to achieve a maximal biological response using concentrated crude COS cell supernatant. However, even at maximal biological concentrations of purified sIL-11R (40 nmol/L), we observed the same requirement for increased IL-11 compared with cells expressing the transmembrane IL-11R.
It is possible that the affinity of IL-11 for the sIL-11R is less than for the transmembrane IL-11R. However, we have estimated the affinity of IL-11 for the sIL-11R is low (10 to 50 nmol/L) and appears similar to that previously measured for the transmembrane IL-11R.13 This is consistent with previous studies of other ligand/soluble receptor interactions. The more probable explanation for the requirement for increased concentration of IL-11 is that once the IL-11/sIL-11R forms, it is less likely, simply on proximity grounds, to encounter a gp130 molecule than an IL-11/transmembrane IL-11R complex (that is, the effective K2 <K3 , Fig 9B). Indeed, the finding that the sIL-11R can compete with the transmembrane IL-11R for IL-11 binding supports this hypothesis. Alternatively, the IL-11/transmembrane complex may truly bind to gp130 with higher affinity than the IL-11/sIL-11R complex. If this is the case, then the transmembrame and/or cytoplasmic domains may be important for stabilization of the ligand/receptor complex.

Model of IL-11 antagonism by sIL-11R. (A) Cells expressing excess gp130-sIL-11R will bind IL-11 and complex with free gp130, thus augmenting the IL-11 response. (B) Cells with limiting gp130 in the presence of limiting IL-11 addition of sIL-11R will compete with the transmembrane IL-11R for IL-11, and antagonism will occur provided K2 <K3 . K1 = affinity of ligand for IL-11R (soluble or transmembrane). K2 = affinity of IL-11/sIL-11R complex for membrane-bound gp130. K3 = affinity of IL-11/transmembrane IL-11R complex for membrane-bound gp130. The + refers to the intensity of the signal generated.
Model of IL-11 antagonism by sIL-11R. (A) Cells expressing excess gp130-sIL-11R will bind IL-11 and complex with free gp130, thus augmenting the IL-11 response. (B) Cells with limiting gp130 in the presence of limiting IL-11 addition of sIL-11R will compete with the transmembrane IL-11R for IL-11, and antagonism will occur provided K2 <K3 . K1 = affinity of ligand for IL-11R (soluble or transmembrane). K2 = affinity of IL-11/sIL-11R complex for membrane-bound gp130. K3 = affinity of IL-11/transmembrane IL-11R complex for membrane-bound gp130. The + refers to the intensity of the signal generated.
The antagonistic activity of sIL-11R was unexpected and has not been previously recognized for recombinant forms of either the sIL-6R or sIL-11R. We believe that the antagonistic activity was real because it was IL-11 specific, seen with all independently purified samples including the monomeric sample (n = 5) and antagonism was observed both for a proliferative and differentiative IL-11 response. The ability of a soluble receptor to behave as an agonist or antagonist has been described for the soluble IL-4 and tumor necrosis factor receptors. However, the agonist activity of these soluble receptors is mediated only via stabilization of their ligands and thus is only seen in cells expressing their transmembrane receptor components.34 35 Why has this activity not been reported previously for the sIL-11R? A possible explanation is the relative number of available gp130 molecules on the cells used to test the activity of sIL-11R. If gp130 is in excess (Fig 9A), then the addition of sIL-11R will provide additional low-affinity complexes capable of binding to gp130 and thus augmenting the IL-11 response. On the other hand, if gp130 is limiting (Fig 9B), then the addition of sIL-11R may be able to compete with the transmembrane IL-11R for gp130. Antagonism of the IL-11 response will then be seen provided the affinity of the IL-11/sIL-11R complex for gp130 (Fig 9B, K2 ) is less than the IL-11/transmembrane IL-11R complex (K3 ). This model correctly predicts that the IL-11 antagonism will be overcome by increasing the concentration of IL-11. It is possible that the greater IL-11 antagonism observed on BaF3/gp130 cells was due to fewer gp130 molecules on BaF3/gp130 cells than M1 cells. Epitope tagging of receptor components would allow an estimation by FACS analysis of relative expressions of gp130 and the IL-11R molecules.
Unlike previous studies, we did not observe any augmentation of the IL-11 response on M1/IL-11R cells in the presence of sIL-11R. However, this is internally consistent with the requirement of greater IL-11 concentrations to signal through the sIL-11R (375 ± 150 pmol/L) compared with the transmembrane IL-11R (15 ± 10 pmol/L). Thus, at a concentration of IL-11 sufficient to signal through the sIL-11R, the M1/IL-11R cells were already maximally stimulated through the transmembrane IL-11R.
Given the existence of soluble receptors for other closely related cytokines and that a transcript potentially encoding a sIL-11R has been described for the mouse,25 it is likely that a naturally occurring sIL-11R exists. However, we have no evidence that a naturally occurring form would behave in a similar fashion to the recombinant form. The physiological functions of soluble receptors are unknown but may include inhibition of the systemic effects of their ligands, stabilization and delivery of ligand to the transmembrane receptor, augmentation of the response in cells expressing the transmembrane receptor, or enabling cells to respond to ligands for which they do not express the specific α-chain receptor.36 The findings presented here suggest an additional degree of complexity which has not been previously recognized with other recombinant soluble receptors: while the sIL-11R can mediate an agonist response, it can compete with the transmembrane IL-11R for IL-11, and thus behave as an IL-11 antagonist. It is possible that this antagonism depends on limiting numbers of gp130 molecules combined with a lower relative affinity of IL-11/sIL-11R complex for gp130 compared with the IL-11/transmembrane IL-11R counterpart. This may have in vivo physiological significance and suggests that the relative numbers of gp130 molecules and specific α-chain receptors on different cell types will need to be assessed.
ACKNOWLEDGMENT
We thank R.J. Simpson for amino acid analysis and L.D. Ward and L. Fabri for biosensor experiments.
Supported in part by grants from the National Health and Medical Research Council (Canberra, Australia); The Anti-Cancer Council of Victoria (Melbourne); and the Cooperative Research Centre for Cellular Growth Factors (Melbourne, Australia).
Address reprint requests to David J. Curtis, MD, Walter and Eliza Hall Institute of Medical Research, Post Office, Royal Melbourne Hospital, Victoria, 3050, Australia.