

Membrane-derived microvesicles (MVs) are released from the cell surface and are implicated in cell-to-cell communication. We evaluated whether MVs derived from endothelial progenitor cells (EPCs) are able to trigger angiogenesis. We found that EPC-derived MVs were incorporated in endothelial cells by interaction with α4 and β1 integrins expressed on the MV surface. In vitro, MVs promoted endothelial cell survival, proliferation, and organization in capillary-like structures. In vivo, in severe combined immunodeficient (SCID) mice, MV-stimulated human endothelial cells organized in patent vessels. When incubated with RNase, despite their internalization into endothelial cells, MVs failed to induce in vitro and in vivo angiogenic effects. mRNA transfer was shown by transduction of GFP protein in endothelial cells by MVs containing GFP-mRNA and the biologic relevance by the angiogenic effect of MV-mRNA extract delivered by lipofectamine. Microarray ana-lysis and quantitative reverse transcription–polymerase chain reaction (RT-PCR) of MV-mRNA extract indicated that MVs were shuttling a specific subset of cellular mRNA, such as mRNA associated with the PI3K/AKT signaling pathway. Protein expression and functional studies showed that PI3K and eNOS play a critical role in the angiogenic effect of MVs. These results suggest that EPCs may activate angiogenesis in endothelial cells by releasing MVs able to trigger an angiogenic program.
Introduction
Stem cells have been proposed as a new opportunity for tissue repair in several diseases. Experimental studies have suggested that transplantation of stem and progenitor cells may have a beneficial effect on functional and structural recovery in several organs, including heart, liver, and kidney.1,–3 The mechanisms underlining stem-cell therapy are still intensely debated. Some studies have suggested an engraftment of stem cells by transdifferentiation or fusion in targeted organs.1,–3 However, a growing number of evidences indicate that transient cell localization in the injured tissue may be sufficient to favor functional and regenerative events, suggesting the release of paracrine mediators.1,–3 Several mechanisms involved in cell-to-cell communication have been identified, including secretion of growth factors, cytokines, surface receptors, and nucleotides.4,,–7 It has been suggested that microvesicles (MVs) actively released from cells may play an important role in cell-to-cell communication.8,,–11
MVs are derived from the endosomal membrane compartment after fusion with the plasma membrane and are shed from the cell surface of activated cells.12,13
Several studies suggest that MVs may stimulate target cells directly or by transferring surface receptors.8,–10,13,14 It has been shown that MVs derived from activated platelets induce metastasis and angiogenesis in lung cancer.14 Moreover, tumor-derived MVs were shown to transfer surface determinants and mRNA of tumor cells to monocytes.15 It has been also postulated that MVs may contribute in spreading certain infective agents such as HIV or prions.16,17
Embryonic stem cells were recently shown to represent an abundant source of MVs, and it was suggested that MVs derived from these cells may represent one of the critical components supporting self-renewal and expansion of stem cells.18,19 In addition, Ratajczak et al18 demonstrated that embryonic stem cell–derived MVs are able to reprogram hematopoietic progenitors by a horizontal transfer of mRNA and protein delivery.
These experimental evidences rise the question whether a stem- cell regenerative therapy is feasible without transplantation of stem cells by using MVs as a carrier of genetic information or proteins able to reprogram tissue resident cells to repair injury.
In the present study, we aimed to investigate whether MVs, derived from human circulating endothelial progenitor cells (EPCs), were able to trigger neoangiogenesis.
Materials and methods
Approval of the study was obtained from the Center for Molecular Biotechnology Institutional Review Board. Adult peripheral blood was collected from healthy volunteers with informed consent obtained in accordance with the Declaration of Helsinki.
Isolation and characterization of EPCs
EPCs were isolated and cultivated as described previously.20,21 In short, EPCs were isolated from peripheral blood mononuclear cells from healthy donors by density centrifugation (Histopaque 1077; Sigma-Aldrich, St Louis, MO). Immediately after isolation, 8 × 106 mononuclear cells/mL medium were plated on fibronectin-coated culture flasks in endothelial cell basal medium-2 (EBM-2; Clonetics Biowhittaker, Walkersville, MD) supplemented with 5% FCS, vascular endothelial growth factor (VEGF), fibroblast growth factor-2, epidermal growth factor, insulinlike growth factor-1, and ascorbic acid.20 After 3 days in culture, nonadherent cells were removed by washing with phosphate-buffered saline (PBS), and cells were cultured in the same medium until colony appearance. When homogeneous monolayers with typical cobblestone morphology were obtained, phenotypic characterization and functional evaluation of angiogenic properties was performed as previously described.20,21 By flow cytometry EPCs were negative for CD2, CD3, CD4, CD5, CD8, CD16, CD20, CD62E, VEGFR-1, CD14, and CD45 but positive for CD34, CD133, Tie-2, VEGFR-3, and VEGFR-2.20 EPCs were characterized by dual-staining for 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine–labeled acetylated low-density lipoprotein and ulex europaeus agglutinin-1 that detects α-L-fucosyl residues on surface glycoproteins expressed on endothelial cells as shown on EPCs20 and lectin and by the expression of endothelial marker proteins VEGFR-2, VE-cadherin, eNOS, and VWF. Endothelial phenotype was further confirmed by Western blot analysis and reverse transcription–polymerase chain reaction (RT-PCR) by expression of markers characteristic for endothelial cells such as Tie-2, VEGFR-2, and VEGFR-3 but not VEGFR-1.20,21 EPCs were transduced with a lentivector carrying a CMV-GFP expression cassette as previously described.22 Briefly, 293T cells were transfected with a 4-plasmid lentiviral system by the CaCl2 precipitation method. Supernatants were collected 48 and 72 hours after transfection, filtered, and concentrated by 2 successive ultracentrifugations. Viral preparation titers were determined by p24 enzyme-linked immunoabsorbent assay (ELISA; Alliance, Perkin-Elmer, Wellesley, MA), by GFP titer determination on 293T cells, and by TaqMan real-time PCR determination of transduced proviral genomes. For gene marking, after 3 to 4 culture passages, EPCs were plated on 24-well plates in complete culture medium and then transduced overnight with a lentivector carrying a CMV-GFP expression cassette, at MOI approximately 20. Culture medium was replaced with fresh medium the following day. EPC transduction was confirmed by fluorescence microscopy and fluorescence-activated cell sorting (FACS) analysis. For this study, EPCs from 10 to 30 passages were used.
Human normal endothelial cell cultures
Human microvascular endothelial cells (HMECs) were isolated from skin specimens of adult healthy persons by magnetic cell sorting using antibody against CD31 (magnetic cell sorting [MACS] system; Miltenyi Biotec, Auburn, CA), cultured, and characterized as previously described.23 Human umbilical vein endothelial cells (HUVECs) were obtained and characterized as described.23
Isolation of MVs
MVs were obtained from supernatants of EPCs cultured in EBM-2 medium deprived of FCS and supplemented with 0.25% BSA. After centrifugation at 2000g for 20 minutes to remove debris, cell-free supernatants were centrifuged at 100 000g (Beckman Coulter Optima L-90K ultracentrifuge; Beckman Coulter, Fullerton, CA) for 1 hour at 4°C, washed in serum-free medium 199 containing N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid 25 mM (Sigma-Aldrich),18 and submitted to a second ultracentrifugation in the same conditions. To trace MVs by fluorescent microscopy or FACS analysis, MVs were labeled with the red fluorescent aliphatic chromophore intercalating into lipid bilayers PKH26 dye (Sigma-Aldrich).24 After labeling, MVs were washed and ultracentrifuged at 100 000g for 1 hour at 4°C. MV pellets were suspended in medium 199, and the protein content was quantified by the Bradford method (BioRad, Hercules, CA). Endotoxin contamination of MVs was excluded by the Limulus test according to the manufacturer's instruction (Charles River Laboratories, Wilmington, MA), and MV were stored at −80°C.
In selected experiments, MVs were treated with 1 U/mL RNase18 (Ambion, Austin, TX) for 1 hour at 37°C, the reaction was stopped by the addition of 10 U/mL RNase inhibitor (Ambion), and MVs were washed by ultracentrifugation. The effectiveness of RNase treatment was evaluated after RNA extraction using TRIZOL reagent (Invitrogen, Carlsbad, CA) by spectrophotometer analysis of total extracted RNA (untreated, 3.3 ± 0.2 μg RNA/mg protein MV; RNase treated, < 0.2 μg RNA/mg protein MV). In addition, RNA extracted from RNase-treated and -untreated MVs was labeled by oligo dT-driven retrotranscription and analyzed on 0.6% Agarose gel to show the complete degradation of RNA by RNase treatment (additional information, Figure 1). As control, MVs were treated with 1 U/mL DNase (Ambion) for 1 hour at 37°C.

Characterization of EPC-derived MVs. (A) Representative FACS analysis of MVs (dark) and of 1-, 2-, 4-, and 6-μm beads used as internal size standards. (B,C) Representative micrographs of scanning electron microscopy of EPC-derived MV showing a spheroid shape. Images were obtained by secondary electron at a working distance of 15 to 25 mm and an accelerating voltage of 20 and 30 kV (original magnification B, ×1500; C, ×3500). Digital acquisition and analysis were performed using the JEOL Semafore system. (D) Representative FACS analysis of MVs showing expression (thick lines) of ICAM-1, α4 integrin, CD44, CD29, αvβ3 integrin, and α6 integrin. Thin lines indicate the isotypic controls. Five MV preparations were analyzed with similar results. In each experiment the Kolmogorov-Smirnov statistical analysis between relevant antibodies and the isotypic control was significant (P < .001).
Characterization of EPC-derived MVs. (A) Representative FACS analysis of MVs (dark) and of 1-, 2-, 4-, and 6-μm beads used as internal size standards. (B,C) Representative micrographs of scanning electron microscopy of EPC-derived MV showing a spheroid shape. Images were obtained by secondary electron at a working distance of 15 to 25 mm and an accelerating voltage of 20 and 30 kV (original magnification B, ×1500; C, ×3500). Digital acquisition and analysis were performed using the JEOL Semafore system. (D) Representative FACS analysis of MVs showing expression (thick lines) of ICAM-1, α4 integrin, CD44, CD29, αvβ3 integrin, and α6 integrin. Thin lines indicate the isotypic controls. Five MV preparations were analyzed with similar results. In each experiment the Kolmogorov-Smirnov statistical analysis between relevant antibodies and the isotypic control was significant (P < .001).
To evaluate the specificity of the angiogenic signal induced by EPC-derived MVs, MVs were also purified from bone marrow–derived human mesenchymal stem cells (MSCs) isolated and characterized as previously described.25
FACS and confocal microscopy analysis
The size of MVs was determined by FACScan (Becton Dickinson Biosciences, San Jose, CA). The instrument was rinsed with particle-free rinse solution for 15 minutes to eliminate the background. The beads of different sizes (1, 2, 4, and 6 μm; Molecular Probes, Invitrogen, Carlsbad, CA) were used as the size markers, and analysis was performed using a log scale for forward scatter and side scatter parameters. The number of MVs diluted in 1:20 ratio was analyzed by flow cytometry during 20-second acquisition of 30 μL in a medium flow option as described by Baj-Krzyworzeka et al.15
Cytofluorimetric analysis was performed as described,23 using the following FITC- or PE-conjugated antibodies (Abs) directed to CD2, CD3, CD4, CD5, CD8, CD16, CD20, CD62E, CD14, CD45, CD34, CD146 (Dako Cytomation, Copenhagen, Denmark); CD133 (Miltenyi Biotec, Bergisch Gladbach, Germany); Tie-2, VEGFR-1, VEGFR-2, VEGFR-3 (R&D System, Minneapolis, MN); ICAM-1, α4 integrin, (Becton Dickinson), αvβ3 integrin, α5 integrin, α6 integrin (BioLegend, San Diego, CA); and soluble Ha (Sigma-Aldrich). FITC or PE mouse nonimmune isotypic IgG (Dako Cytomation) was used as control. Indirect immunofluorescence was performed on cells cultured on chamber slides, fixed in 4% paraformaldehyde containing 2% sucrose.21 Confocal microscopy was performed using a Zeiss confocal microscope, model LSM 5 PASCAL (Jena, Germany). Anti–human HLA class I antigen was from Santa Cruz Biotechnology (Santa Cruz, CA); anti–human CD31 Ab was from Becton Dickinson. Hoechst 33258 dye (Sigma-Aldrich) was added for nuclear staining.
Scanning electron microscopy
MVs were fixed in Karnowski fixative, dehydrated in alcohol, dried on glass surface, and coated with gold by sputter coating. The specimens were examined in a scanning Jeol T300 electron microscope (Tokyo, Japan). Images were obtained by secondary electron at a working distance of 15 to 25 mm and an accelerating voltage of 20 to 25 kV.
Cell proliferation and apoptosis assays
Cells were seeded at 8000 cells/well into 96-well plates in EBM-2 deprived of FCS and endothelial growth factors. DNA synthesis was detected as incorporation of 5-bromo-2′-deoxy-uridine (BrdU) into the cellular DNA using an ELISA kit (Roche Applied Science, Mannheim, Germany), following the manufacturer's instructions.23 Apoptosis was evaluated using the terminal dUTP nick-end labeling (TUNEL) assay analysis (ApopTag Oncor, Gaithersburg, MD) as previously described.23
Western blot analysis
Cells were lysed at 4°C for 1 hour in a lysis buffer (50 mM Tris-HCl, pH 8.3, containing 1% Triton X-100, 1 mM PMSF, 10 μg/mL leupeptin, and 100 U/mL aprotinin). Aliquots of the cell lysates containing 30 μg protein, as determined by the Bradford method, were subjected to 4% to 15% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions and electroblotted onto nitrocellulose membrane filters as previously described.26 The following primary antibodies were used: mAb against Akt/PKB (Upstate, Charlottesville, VA), mAb against phospho-Akt and rabbit polyclonal Ab against phospho-eNOS (Cell Signaling, Beverly, MA), mAb against actin and Bcl-xL and rabbit polyclonal Ab against eNOS (Santa Cruz Biotechnology).
In vitro angiogenesis
In vitro formation of capillary-like structures was studied on HMECs and HUVECs (5 × 104 cells/well) seeded on growth factor–reduced Matrigel (Becton Dickinson) diluted 1:1 in ice with cold Dulbecco Modified Eagle Medium (DMEM; Sigma-Aldrich).23 After cells had attached, the medium was removed and 1 mL medium containing EPC-derived MVs was added. Cells were observed under a Nikon-inverted microscope (Kanagawa, Japan), using a 10×/0.25 NA objective lens, and experimental results were recorded after 6-hour incubation at 37°C. Image analysis was performed with the MicroImage analysis system (Casti Imaging, Venice, Italy).23 In selected experiments 10 μg/mL blocking Abs anti-αvβ3 integrin (BioLegend, San Diego, CA), α4 integrin, α5 integrin (Chemicon, Temecula, CA) or CD29 (Becton Dickinson Biosciences) were added to MV-stimulated HMECs.
In vivo angiogenesis
For the in vivo studies of MV-induced angiogenesis, HMECs were first incubated with MVs or RNase-treated MVs for 30 minutes at 37°C and then implanted subcutaneously into severe combined immunodeficient (SCID) mice (Charles River Laboratories; n = 6 for each experimental condition) within growth factor–reduced Matrigel as previously described.23,27 At day 7, mice were killed, and Matrigel plugs were recovered and processed for histology. Sections stained with hematoxylin and eosin or with a Masson tricromic reaction were examined under a light microscopy. Vessels were counted only if showing a patent lumen with erythrocytes, leukocytes, or both. The vessel area was planimetrically assessed as percentage area per field using the MicroImage analysis system (Casti Imaging). The human nature of vessels was assessed by immunofluorescence staining for human HLA class I antigen and CD31, as previously described.23
Gene array analysis
RNA extraction, samples labeling, and hybridization on BeadChips.
RNA was extracted from MVs using TRIZOL reagent (Invitrogen) following the procedure suggested by the manufacturer. Total RNA was quantified spectrophotometrically. cRNA was synthesized using 3 different quantities of total RNA (250 ng, 1 μg, and 2.5 μg). cRNA synthesis and labeling were done using Illumina RNA Amplification Kit (Ambion) following the procedure suggested by the manufacturer. Sentrix Human-6 Expression BeadChip hybridization, washing, and staining were also done as suggested by the manufacturer. Arrays were scanned on Illumina BeadStation 500 (Illumina, San Diego, CA).
Microarray data analysis.
BeadChip array data quality control was performed using Illumina BeadStudio software version 1.3.1.5. Transcript average-intensity signals were calculated with BeadStudio without background correction. Raw data were analyzed using Bioconductor.28 Average transcript intensities were log2 transformed and normalized by loess method.29
A simple statistical linear model was used to identify transcript signals linearly correlated to the increment of total RNA concentration used to prepare cRNA. In equation (1) yij is the observed expression level for transcript i in sample j (j = 1,… ,6); μ is the average expression level of transcript i and βRNA represents the effect of total RNA concentration on the expression level of transcript i. ϵ represents random error for transcript i and sample j, and it is assumed to be independent for each transcript and sample, and normally distributed with mean 0 and variance σ2.
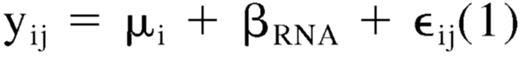
Transcripts characterized by a model with P ≤ .05, r2 ≥ .8, and a positive slope were selected (298).
Transcript annotation and data mining were performed using IPA 4.0 software.30
Microarray data were deposited on GEO database (http://www.ncbi.nlm.nih.gov/projects/geo/) as GSE7019 series.
Quantitative RT real-time PCR (qPCR).
qPCR was performed on total RNA extracted from cells used to produce MVs and from a MV preparation different from those used for microarray analysis. Cell cDNA concentration curve was used to evaluate primer efficiency. Total RNA was reverse-transcribed to cDNA using Omniscript RT Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. Primers (Table 1) were designed by Primer Express 2.0 software (Applied Biosystems, Foster City, CA), using the reference sequence (RefSeq) associated to each BeadChip identifier. qPCR mix (20 μL), containing 1X SYBR GREEN PCR Master Mix (Applied Biosystems), 150 nM of each primer and, respectively, 0 μL, 0.25 μL, 1 μL, 2.5 μL of MV cDNA, were assembled on 384 plates by QIAGEN 8000 BIOROBOT (Qiagen).
Primers used for qPCR validation
| Gene | Primer FW | Primer RW |
|---|---|---|
| ACTB | GAGTCCGGCCCCTCCAT | GCAACTAAGTCATAGTCCGCCTAGA |
| BCL2L1 | TCAGTCGGAAATGACCAGACACT | GGATGTGGTGGAGCAGAGAAG |
| CFL1 | CAAGGACGCCATCAAGAAGAA | ACCTCCTCGTAGCAGTTTGCTT |
| CTNNB1 | GCTGGCCTGGTTTGATACTGA | GTAAAAGTATTTTACCCAAACTGGCTTT |
| EDF1 | TTGGAAAGCCCATCGAGAAG | GCACTGATTTCGAGGCTTTGT |
| MAPKAPK2 | GGAGCGGTGGGAGGATGT | CGTAGTCAACGCGCATTGTG |
| POLR2B | CCTGATCATAACCAGTCCCCTAGA | GTAAACTCCCATAGCCTGCTTACC |
| PTPRT | GGGCGTGATGCTCACCAT | CTTTGGGACAAGTAATAGGAGTAGGAA |
| eNOS | CGT GGG CCG GAT CCA | GTG GTT GCA GAT GTA GGT GAA CA |
| Gene | Primer FW | Primer RW |
|---|---|---|
| ACTB | GAGTCCGGCCCCTCCAT | GCAACTAAGTCATAGTCCGCCTAGA |
| BCL2L1 | TCAGTCGGAAATGACCAGACACT | GGATGTGGTGGAGCAGAGAAG |
| CFL1 | CAAGGACGCCATCAAGAAGAA | ACCTCCTCGTAGCAGTTTGCTT |
| CTNNB1 | GCTGGCCTGGTTTGATACTGA | GTAAAAGTATTTTACCCAAACTGGCTTT |
| EDF1 | TTGGAAAGCCCATCGAGAAG | GCACTGATTTCGAGGCTTTGT |
| MAPKAPK2 | GGAGCGGTGGGAGGATGT | CGTAGTCAACGCGCATTGTG |
| POLR2B | CCTGATCATAACCAGTCCCCTAGA | GTAAACTCCCATAGCCTGCTTACC |
| PTPRT | GGGCGTGATGCTCACCAT | CTTTGGGACAAGTAATAGGAGTAGGAA |
| eNOS | CGT GGG CCG GAT CCA | GTG GTT GCA GAT GTA GGT GAA CA |
qPCR analysis was done on ABI PRISM 7900HT Sequence Detection System, using the following conditions: 50°C for 2 minutes, 95°C for 10 minutes, and 95°C for 15 seconds followed by 40 cycles at 60°C for 1 minute. Negative cDNA controls (no cDNA) were cycled in parallel with each run. Fluorescence data were analyzed with the SDS 2.1 software (Applied Biosystems) and expressed as Ct (ie, the number of cycles needed to generate a fluorescent signal above a predefined threshold).
Results
Characterization of EPC-derived MVs
FACS analysis of MVs derived from human EPCs was used to determine their size. Using 1-, 2-, 4-, and 6-μm beads as size internal standards, the majority of MVs were observed around the forward scatter signal corresponding to 1-μm beads (Figure 1A). Scanning electron microscopy showed the spheroid morphology of MVs and confirmed that their size was approximately 1 μm (Figure 1B,C). The number of events recorded by FACS analysis was 10 785 (± 521; mean ± SD) per 1 μg protein of MVs.
FACS analysis showed the expression by MVs of several adhesion molecules known to be present on EPC plasma membrane such as intracellular adhesion molecule-1 (ICAM-1), α4 integrin, CD44, and CD29 (β1 integrin) but not ανβ3 integrin and α6 integrin adhesion molecules (Figure 1D). In addition, MVs bound ulex europaeus agglutinin-1 which is known to interact with selectin ligands expressed by EPCs and endothelial cells (not shown).20,21 These results indicate that MVs express on their surface several adhesion molecules of plasma membranes of EPCs from which they originated. The expression of adhesion molecules may be instrumental in the incorporation of MVs in target cells.
Incorporation of EPC-derived MVs in endothelial cells
MVs labeled with PKH26 dye were incorporated by HMECs after a 30-minute incubation at 37°C as shown by FACS analysis (Figure 2A) and confocal microscopy (not shown). To investigate the role of adhesion molecules expressed by the MV surface in the incorporation in target cells, MVs were preincubated (15 minutes at 4°C) with blocking Abs against the identified adhesion molecules (Figure 2B-F). MV treatment with anti-α4 integrin and anti-CD29 blocking Abs inhibited MV incorporation in HMECs, suggesting that the expression of these molecules is instrumental in MV internalization (Figure 2E,F). In contrast, blockade of ICAM-1, α6 integrin with specific Abs, or CD44 with soluble hyaluronic acid (Ha) did not prevent internalization of MVs (Figure 2B-D). Similar results were obtained with HUVECs (not shown).

Incorporation of EPC-derived MVs in HMECs. (A) Representative FACS analysis of internalization, after a 30-minute incubation at 37°C, by HMECs of MVs labeled with PKH26 (filled curve). HMECs were incubated with vehicle alone as internal control (open curve). (B-F) Representative FACS analysis of internalization of MVs preincubated (open curves) or not (filled curves) with 1 μg/mL blocking mAb against ICAM-1 (B), with 5 μg/mL soluble Ha (C), 1 μg/mL blocking mAb against α6 integrin (D), α4 integrin (E), CD29 (F). Filled curves indicate the MVs internalized in the absence of blocking Abs or Ha; open curves indicate the MVs internalized after incubation with blocking Abs or soluble Ha. Three experiments were performed with similar results.
Incorporation of EPC-derived MVs in HMECs. (A) Representative FACS analysis of internalization, after a 30-minute incubation at 37°C, by HMECs of MVs labeled with PKH26 (filled curve). HMECs were incubated with vehicle alone as internal control (open curve). (B-F) Representative FACS analysis of internalization of MVs preincubated (open curves) or not (filled curves) with 1 μg/mL blocking mAb against ICAM-1 (B), with 5 μg/mL soluble Ha (C), 1 μg/mL blocking mAb against α6 integrin (D), α4 integrin (E), CD29 (F). Filled curves indicate the MVs internalized in the absence of blocking Abs or Ha; open curves indicate the MVs internalized after incubation with blocking Abs or soluble Ha. Three experiments were performed with similar results.
Proliferative and antiapoptotic effects of EPC-derived MVs
Incubation of HMECs with different doses of MVs derived from EPCs promoted cell proliferation in respect to control cells incubated with vehicle alone. The proliferation was significant at the dose of 10 μg protein MV/mL (Figure 3A). As shown in Figure 3B, a similar proliferative effect was evident in HUVECs stimulated with 10 μg/mL MVs. In addition, incubation of HMECs with increasing doses of MVs significantly inhibited apoptosis induced by serum deprivation (Figure 3C). The antiapoptotic effect was present also in HUVECs stimulated with 10 μg/mL MVs (Figure 3D). MV treatment with anti-α4 integrin and anti-CD29 blocking Abs, that inhibited MV incorporation, or with both, also inhibited the proliferative and antiapoptotic effect on HUVECs (Figure 3B-D), suggesting that MV incorporation was required for the activity of MVs. However, when MVs were incubated with RNase, the proliferation and the antiapoptotic effects elicited by MVs were significantly reduced (Figure 3A-D), suggesting that these biologic effects were mediated by the transfer of mRNA after MV internalization. DNase treatment was ineffective (Figure 3B). Before cell stimulation, RNase was inhibited by incubation with RNase inhibitor and removed by washing. However, to exclude that residual RNase associated with MVs may interfere with the endothelial proliferation, VEGF-induced (10 ng/mL) proliferation was evaluated in HMECs preincubated with RNase-treated MVs (average OD intensity, 1206 ± 80.23) or not (average OD intensity, 1130 ± 59.65), and no differences in cell proliferation was observed, suggesting that RNase did not interfere with this endothelial cell function.

Proliferative and antiapoptotic effects of EPC-derived MVs. (A,B) Eight thousand cells/well (A, HMECs; B, HUVECs) into 96-well plates were added with 10 μM BrdU, incubated for 72 hours in EBM-2 without FCS and growth factors in the presence of vehicle alone or of different doses of MVs (A, HMECs ■) or with 1 U/mL RNase-treated MVs (□). HUVECs (B) were incubated with vehicle alone (M199 medium) or vehicle plus 1 μg/mL of anti-α4 integrin– and anti-CD29–blocking Abs or with 10 μg/mL MVs alone or 10 μg/mL MVs preincubated (30 minutes, 37°C) with 1 μg/mL anti-α4 integrin or anti-CD29–blocking Ab or both or with 1 U/mL RNase or DNase (1 hour, 37°C). Cells were then fixed with 0.5 M ethanol/HCl and incubated with nuclease to digest the DNA. BrdU incorporated into the DNA was detected using an anti-BrdU peroxidase-conjugated mAb and visualized with a soluble chromogenic substrate. Optical density was measured with an ELISA reader at 405 nm. Results are expressed as mean (± 1 SD) of 3 experiments. Analysis of variance with Newmann-Keuls multicomparison test was performed; §P < .05 MVs versus vehicle alone; *P < .05 MV treatments versus MV alone. (C,D) The percentage of apoptotic cells after 48-hour serum withdrawal was evaluated by the TUNEL assay. HMECs (C) were incubated with vehicle alone or with different doses of MVs (■) or with 1 U/mL RNase-treated MV (▨). HUVECs (D) were incubated with vehicle alone or vehicle plus 1 μg/mL of anti-α4 integrin and anti-CD29–blocking Abs or with 10 μg/mL MV alone or 10 μg/mL MV preincubated (30 minutes, 37°C) with 1 μg/mL anti-α4 integrin or anti-CD29–blocking Ab or both or with 1 U/mL RNase (1 hour, 37°C). Results are expressed as mean (± 1 SD) of 3 experiments. Analysis of variance with Newmann-Keuls multicomparison test was performed; *P < .05 MVs versus vehicle alone; §P < .05 MV treatments versus MVs alone.
Proliferative and antiapoptotic effects of EPC-derived MVs. (A,B) Eight thousand cells/well (A, HMECs; B, HUVECs) into 96-well plates were added with 10 μM BrdU, incubated for 72 hours in EBM-2 without FCS and growth factors in the presence of vehicle alone or of different doses of MVs (A, HMECs ■) or with 1 U/mL RNase-treated MVs (□). HUVECs (B) were incubated with vehicle alone (M199 medium) or vehicle plus 1 μg/mL of anti-α4 integrin– and anti-CD29–blocking Abs or with 10 μg/mL MVs alone or 10 μg/mL MVs preincubated (30 minutes, 37°C) with 1 μg/mL anti-α4 integrin or anti-CD29–blocking Ab or both or with 1 U/mL RNase or DNase (1 hour, 37°C). Cells were then fixed with 0.5 M ethanol/HCl and incubated with nuclease to digest the DNA. BrdU incorporated into the DNA was detected using an anti-BrdU peroxidase-conjugated mAb and visualized with a soluble chromogenic substrate. Optical density was measured with an ELISA reader at 405 nm. Results are expressed as mean (± 1 SD) of 3 experiments. Analysis of variance with Newmann-Keuls multicomparison test was performed; §P < .05 MVs versus vehicle alone; *P < .05 MV treatments versus MV alone. (C,D) The percentage of apoptotic cells after 48-hour serum withdrawal was evaluated by the TUNEL assay. HMECs (C) were incubated with vehicle alone or with different doses of MVs (■) or with 1 U/mL RNase-treated MV (▨). HUVECs (D) were incubated with vehicle alone or vehicle plus 1 μg/mL of anti-α4 integrin and anti-CD29–blocking Abs or with 10 μg/mL MV alone or 10 μg/mL MV preincubated (30 minutes, 37°C) with 1 μg/mL anti-α4 integrin or anti-CD29–blocking Ab or both or with 1 U/mL RNase (1 hour, 37°C). Results are expressed as mean (± 1 SD) of 3 experiments. Analysis of variance with Newmann-Keuls multicomparison test was performed; *P < .05 MVs versus vehicle alone; §P < .05 MV treatments versus MVs alone.
In vitro and in vivo proangiogenic effect of EPC-derived MVs
In preliminary experiments HMECs or HUVECs were incubated with different doses of MVs derived from EPCs (1-30 μg protein MV/mL), and it was found a significant in vitro angiogenic response at the dose of 10 μg protein MV/5 × 104 cells/mL corresponding to an approximate number of 2.1 MVs per target cell. Stimulation of HMECs or HUVECs with 10 μg/mL of MVs promoted their organization into capillary-like structures on Matrigel after a 6-hour incubation at 37°C (Figure 4A,B). In respect to control incubated with vehicle alone, HMECs and HUVECs incubated with MVs showed an enhanced formation of ringlike structures, suggesting that MVs stimulated organization and cell-to-cell interaction required for the in vitro formation of capillary-like structures (Figure 4A,B). When MVs were incubated with RNase but not with DNase, the capillary-like formation was significantly reduced (Figure 4A,B). RNase-MVs did not interfere with the ability of endothelial cells to form capillary-like structures when stimulated with 10 ng/mL VEGF (Figure 4A). Moreover, the specificity of EPC-derived MVs to trigger the angiogenic signal was indicated by the absence of capillary-like formation after stimulation with MVs derived from MSCs (Figure 4A).

In vitro proangiogenic effects of EPC-derived MVs. (A) HMECs (■) or HUVECs (□) (5 × 104 cells/well) were plated on growth factor–reduced Matrigel in DMEM + 0.25% BSA and were challenged for 6 hours at 37°C with vehicle alone (M199 medium) as control or with 10 μg protein EPC-derived MVs/mL, or RNase- or DNase (1 U/mL)–treated MVs, or 10 ng/mL VEGF or 10 ng/mL VEGF in the presence of RNase-treated MVs. In selected experiments, HMECs were stimulated with 10 μg protein MSC-derived MVs/mL. Data show the mean (± 1 SD) of total length of capillary-like structures detected by Nikon Eclipse TE 200 inverted microscope (objective, 10×/0.25; Tokyo, Japan), analyzed by the Micro-Image system (Casti Imaging) and expressed as arbitrary units by the computer analysis system in 5 different files at × 100 magnification in duplicated wells of 4 different experiments. Analysis of variance with Newmann-Keuls multicomparison test was performed; §P < .05 different treatments versus unstimulated control; *P < .05 RNase MVs and MSC-derived MVs versus EPC-derived MVs. (B) Representative micrographs of capillary-like structure formation on Matrigel by HMECs unstimulated (Control) and stimulated with either 10 μg/mL MVs or with RNase(1 U/mL)–treated MVs (scale bar, 20 μm). (C) Representative FACS analysis of ανβ3 and α5 integrin expression by HMECs unstimulated (gray filled curves) or stimulated with 10 μg/mL MVs (open curves, dark lines). Dotted lines indicate the isotypic controls. Two MV preparations were analyzed with similar results. In each experiment the Kolmogorov-Smirnov statistical analysis between relevant antibodies and the isotypic control was significant (P < .001). (D) Effect of 10 μg/mL blocking antibodies anti-ανβ3 and anti-α5 integrins on capillary-like structure formation by HMECs cultured on Matrigel added 30 minutes after stimulation with 10 μg/mL MVs. Analysis of variance with Newmann-Keuls multicomparison test was performed; *P < .05 MV plus blocking antibodies versus MVs alone.
In vitro proangiogenic effects of EPC-derived MVs. (A) HMECs (■) or HUVECs (□) (5 × 104 cells/well) were plated on growth factor–reduced Matrigel in DMEM + 0.25% BSA and were challenged for 6 hours at 37°C with vehicle alone (M199 medium) as control or with 10 μg protein EPC-derived MVs/mL, or RNase- or DNase (1 U/mL)–treated MVs, or 10 ng/mL VEGF or 10 ng/mL VEGF in the presence of RNase-treated MVs. In selected experiments, HMECs were stimulated with 10 μg protein MSC-derived MVs/mL. Data show the mean (± 1 SD) of total length of capillary-like structures detected by Nikon Eclipse TE 200 inverted microscope (objective, 10×/0.25; Tokyo, Japan), analyzed by the Micro-Image system (Casti Imaging) and expressed as arbitrary units by the computer analysis system in 5 different files at × 100 magnification in duplicated wells of 4 different experiments. Analysis of variance with Newmann-Keuls multicomparison test was performed; §P < .05 different treatments versus unstimulated control; *P < .05 RNase MVs and MSC-derived MVs versus EPC-derived MVs. (B) Representative micrographs of capillary-like structure formation on Matrigel by HMECs unstimulated (Control) and stimulated with either 10 μg/mL MVs or with RNase(1 U/mL)–treated MVs (scale bar, 20 μm). (C) Representative FACS analysis of ανβ3 and α5 integrin expression by HMECs unstimulated (gray filled curves) or stimulated with 10 μg/mL MVs (open curves, dark lines). Dotted lines indicate the isotypic controls. Two MV preparations were analyzed with similar results. In each experiment the Kolmogorov-Smirnov statistical analysis between relevant antibodies and the isotypic control was significant (P < .001). (D) Effect of 10 μg/mL blocking antibodies anti-ανβ3 and anti-α5 integrins on capillary-like structure formation by HMECs cultured on Matrigel added 30 minutes after stimulation with 10 μg/mL MVs. Analysis of variance with Newmann-Keuls multicomparison test was performed; *P < .05 MV plus blocking antibodies versus MVs alone.
After stimulation with 10 μg/mL MV, HMECs enhanced the expression of ανβ3 and α5 integrins (Figure 4C) but not of α6 integrin and CD29 (not shown). When HMECs were stimulated with MVs and after 30 minutes treated with blocking Abs against ανβ3 and α5 integrins (Figure 4D) but not against α6 integrin and CD29 (not shown), MV-induced angiogenesis was significantly inhibited.
In vivo, when HMECs preincubated for 30 minutes at 37°C with 30 μg/mL MV derived from EPCs were injected subcutaneously within Matrigel in SCID mice, they organized after 7 days in patent vessels (Figure 5A) of variable size connected with the murine vasculature as shown by the presence of erythrocytes in the lumen (Figure 5B). The human nature of implanted endothelial cells was assessed by immunofluorescence staining for human HLA class I and human CD31 (Figure 5C). Control HMECs injected with vehicle alone (Figure 5A,B) or HMECs injected with RNase-treated MVs (Figure 5A,B) did not form vessels within Matrigel.

In vivo proangiogenic effects of EPC-derived MV. Formation of vessels after 7 days by HMECs preincubated (30 minutes at 37°C) with 30 μg/mL of MVs and then injected subcutaneously in Matrigel in SCID mice. (A) Quantitative evaluation of angiogenesis induced by MVs or by RNase-treated MVs. Angiogenesis was evaluated as the percentage of vessel area, and data are expressed as mean (± 1 SD) of 6 experiments. Analysis of variance with Newmann-Keuls test was performed; *P < .05 MVs versus control; §P < .05 RNase MVs versus MVs. (B) Representative micrographs of hematoxylin and eosin staining of Matrigel plugs obtained by Zeiss Axioskop (Jena, Germany) using ×100 and ×250 objectives. We considered as patent vessels those connected with the murine vasculature as shown by the presence of erythrocytes in the lumen (arrows). (Scale bar, 20 μm). (C) Representative confocal micrographs (Zeiss LSM 5 Pascal confocal Laser scanning microscope equipped with an Helium/Neon 543 mm laser, an Argon 450-530 mm laser, and an EC planar NEOFluar 40×/1.3 oil DIC objective lens; acquisition software, Zeiss LSMS version 3.2) showing the expression of human HLA class I antigen and human CD31 by HMEC-formed vessels within the implanted Matrigel (scale bar, 10 μm).
In vivo proangiogenic effects of EPC-derived MV. Formation of vessels after 7 days by HMECs preincubated (30 minutes at 37°C) with 30 μg/mL of MVs and then injected subcutaneously in Matrigel in SCID mice. (A) Quantitative evaluation of angiogenesis induced by MVs or by RNase-treated MVs. Angiogenesis was evaluated as the percentage of vessel area, and data are expressed as mean (± 1 SD) of 6 experiments. Analysis of variance with Newmann-Keuls test was performed; *P < .05 MVs versus control; §P < .05 RNase MVs versus MVs. (B) Representative micrographs of hematoxylin and eosin staining of Matrigel plugs obtained by Zeiss Axioskop (Jena, Germany) using ×100 and ×250 objectives. We considered as patent vessels those connected with the murine vasculature as shown by the presence of erythrocytes in the lumen (arrows). (Scale bar, 20 μm). (C) Representative confocal micrographs (Zeiss LSM 5 Pascal confocal Laser scanning microscope equipped with an Helium/Neon 543 mm laser, an Argon 450-530 mm laser, and an EC planar NEOFluar 40×/1.3 oil DIC objective lens; acquisition software, Zeiss LSMS version 3.2) showing the expression of human HLA class I antigen and human CD31 by HMEC-formed vessels within the implanted Matrigel (scale bar, 10 μm).
Evidence for mRNA transfer by EPC-derived MVs and role of mRNA in mediating the proangiogenic effect
The transfer of mRNA from EPCs to HMECs by MVs was demonstrated using MVs derived from EPCs transduced with a lentivector carrying a CMV-GFP expression cassette. MVs derived from GFP-EPCs did not contain GFP proteins in an amount sufficient to be detected by confocal microscopy (Figure 6B). HMECs incubated with MVs derived from GFP-EPCs started to synthesize GFP which became detectable by confocal microscopy after a 6-hour incubation (Figure 6E), increased after 24 hours (Figure 6F), to decrease thereafter (Figure 6G). Treatment with RNase abrogated the expression of GFP by HMECs incubated with MVs derived from GFP-EPCs (Figure 6H) despite their internalization (Figure 6I).

mRNA transfer by EPC-derived MVs. HMECs (5 × 104 cells/well) were incubated for 24 hours in DMEM plus 5% BSA with 10 μg/mL MVs derived from GFP-EPC, and the synthesis of GFP protein was evaluated by confocal microscopy (Zeiss LSM 5 Pascal confocal Laser scanning microscope equipped with an Helium/Neon 543 mm laser, an Argon 450-530 mm laser, and an EC planar NEOFluar 63 × 1.4 oil DIC objective lens; acquisition software, Zeiss LSMS version 3.2). (A) Representative micrograph showing MVs labeled with the PKH26 red fluorescent dye. (B) Representative micrograph showing that the green fluorescence of GFP was not detectable by confocal microscopy in MVs derived from GFP-EPC. (C) Representative micrograph showing the uptake of PKH26-labeled MVs by HMECs after a 30-minute incubation at 37°C. (D-G) The detection is shown of green fluorescence in HMECs incubated with MVs derived from GFP-EPC after 30 minutes (D), 6 hours (E), 24 hours (F), or 72 hours (G) at 37°C. (H) Treatment with RNase abrogated the expression of GFP by HMECs incubated for 24 hours at 37°C with MVs derived from GFP-EPCs despite that labeled MVs were internalized by HMECs (I). Three experiments were performed with similar results. Scale bar, 10 μm. (J-M) Effect of MV mRNA extracts on in vitro angiogenesis assay. HMECs (5 × 104 cells/well) were plated on growth factor–reduced Matrigel in DMEM plus 0.25% BSA and were challenged with vehicle alone or were stimulated with 10 μg/mL MVs or with 6μg Lipofectamine 2000 (Invitrogen) alone (Lf) or Lf plus 3 μg MV mRNA extracts, 3 μg mRNA alone, and 1 U/mL Rnase plus Lf plus MV mRNA extracts for 6 hours at 37°C. (J) Data show the mean (± 1 SD) of total length of capillary-like structures expressed as arbitrary units by the computer analysis system in 5 different files at ×20 magnification in duplicated wells of 4 different experiments. Analysis of variance with Newmann-Keuls multicomparison test was performed; *P < .05 treatments versus vehicle; §P < .05 mRNA alone or Lf plus mRNA plus RNase versus Lf plus mRNA extracts. (K-M) Representative micrographs showing the capillary-like structure formation on Matrigel by HMECs unstimulated (K) and stimulated with either (L) Lf + mRNA extracted from MVs or (M) with Lf + mRNA + RNase (Scale bar, 20 μm). The images were obtained by Nikon inverted microscope coupled with Casti Microimage analysis system as described in “In vitro angiogenesis.”
mRNA transfer by EPC-derived MVs. HMECs (5 × 104 cells/well) were incubated for 24 hours in DMEM plus 5% BSA with 10 μg/mL MVs derived from GFP-EPC, and the synthesis of GFP protein was evaluated by confocal microscopy (Zeiss LSM 5 Pascal confocal Laser scanning microscope equipped with an Helium/Neon 543 mm laser, an Argon 450-530 mm laser, and an EC planar NEOFluar 63 × 1.4 oil DIC objective lens; acquisition software, Zeiss LSMS version 3.2). (A) Representative micrograph showing MVs labeled with the PKH26 red fluorescent dye. (B) Representative micrograph showing that the green fluorescence of GFP was not detectable by confocal microscopy in MVs derived from GFP-EPC. (C) Representative micrograph showing the uptake of PKH26-labeled MVs by HMECs after a 30-minute incubation at 37°C. (D-G) The detection is shown of green fluorescence in HMECs incubated with MVs derived from GFP-EPC after 30 minutes (D), 6 hours (E), 24 hours (F), or 72 hours (G) at 37°C. (H) Treatment with RNase abrogated the expression of GFP by HMECs incubated for 24 hours at 37°C with MVs derived from GFP-EPCs despite that labeled MVs were internalized by HMECs (I). Three experiments were performed with similar results. Scale bar, 10 μm. (J-M) Effect of MV mRNA extracts on in vitro angiogenesis assay. HMECs (5 × 104 cells/well) were plated on growth factor–reduced Matrigel in DMEM plus 0.25% BSA and were challenged with vehicle alone or were stimulated with 10 μg/mL MVs or with 6μg Lipofectamine 2000 (Invitrogen) alone (Lf) or Lf plus 3 μg MV mRNA extracts, 3 μg mRNA alone, and 1 U/mL Rnase plus Lf plus MV mRNA extracts for 6 hours at 37°C. (J) Data show the mean (± 1 SD) of total length of capillary-like structures expressed as arbitrary units by the computer analysis system in 5 different files at ×20 magnification in duplicated wells of 4 different experiments. Analysis of variance with Newmann-Keuls multicomparison test was performed; *P < .05 treatments versus vehicle; §P < .05 mRNA alone or Lf plus mRNA plus RNase versus Lf plus mRNA extracts. (K-M) Representative micrographs showing the capillary-like structure formation on Matrigel by HMECs unstimulated (K) and stimulated with either (L) Lf + mRNA extracted from MVs or (M) with Lf + mRNA + RNase (Scale bar, 20 μm). The images were obtained by Nikon inverted microscope coupled with Casti Microimage analysis system as described in “In vitro angiogenesis.”
To evaluate whether mRNA derived from MVs was the main effector of proangiogenic stimulation independently from internalization of MVs mediated by adhesion molecules, HMECs were stimulated with mRNA extracted from MVs in the presence of lipofectamine. As shown in Figure 6J and L, MV mRNA stimulated a significant organization of HMECs into capillary-like structures on Matrigel after a 6-hour incubation at 37°C. Lipofectamine as well as mRNA alone or treatment with RNase of mRNA extracted from MVs was ineffective (Figure 6J,K,M).
Gene array analysis of MVs
To identify the mRNA species shuttled by the microvesicles we used microarray analysis. Specifically, microarrays were used not to define the amount of mRNA in the MVs but only to define which transcripts were present. Transcripts present in the MVs were defined as those characterized by a positive linear relation between the transcript signal detected by the microarray analysis and the amount of total RNA hybridized. This analysis was done using hybridizing arrays with labeled cRNA produced using 3 different concentrations of total RNA extracted from 2 independent preparations of MVs. A total of 298 transcripts were found with this procedure, 183 were associated to RefSeq identifiers31 and the remaining were Unigene EST (Table S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). This observation indicates that MVs are not shuttling a random sample of cellular mRNA but a specific subset. To understand the presence of functional relations existing between MV mRNA we took advantage of Ingenuity database,30 the world's largest database of biologic networks. Seventy-eight of 183 RefSeq were eligible for network generation and 6 networks could be created. Because of the limited number of transcripts available for this analysis, literature networks were had little information (not shown). However, looking at the known pathways available in Ingenuity, we observed that 4 (IKK; β-catenine; BCL-XL; eNOS) of the 78 transcripts were associated to the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway (Figure S2).
Quantitative RT-PCR was used to confirm the presence in the MVs of relevant genes (BCL-XL, CFL1, CTNNB1, EDF1, MAPKAPK2, PTPRT, eNOS). Interestingly, one of the genes used as reference in the qPCR analysis, POLR2B, was shown to be present in the MVs. This gene encodes the second largest subunit of RNA polymerase II, the polymerase responsible for synthesizing messenger RNA in eukaryotes.
MVs induced AKT activation and expression of eNOS
The activation of Akt survival pathway by MVs was indicated by a significant increase of Akt phosphorylation in serum-starved HMECs incubated with 10 μg/mL MVs for 24 hours. The enhanced Akt phosphorylation was abrogated by treatment of MVs with RNase (Figure 7A,B). Moreover, incubation with MVs induced expression of Bcl-xL and of e-NOS by HMECs (Figure 7B,C). The activation of eNOS was indicated by its phosphorylation at ser1177 (Figure 7C). As shown in Figure 7D, the activation of Akt-dependent pathway and of eNOS was critical for the in vitro angiogenic effect of MVs because the organization of HMECs into capillary-like structures on Matrigel was inhibited by PI3K inhibitors wortmannin (0.1 μM) and LY294002 (10 μM)32 and by the NOS inhibitors NG-nitro-L-arginine-methyl ester (L-NAME; 10 μM) and NG-monomethyl-L-arginine (L-NMMA; 10 μM).33 Their inactive enantiomers, D-NAME and D-NMMA, were ineffective.

MVs induced Akt activation and expression of Bcl-xL and eNOS. Cell lysates (30 μg of protein) were immunoblotted with anti–P-Akt, -Akt, – Bcl-xL or -βactin Abs. (A) Densitometric analysis of P-Akt/Akt ratio. Data of P-Akt/Akt ratio are expressed as mean (± 1 SD) from 3 different experiments. (B) Representative immunoblots of P-Akt, Akt, and Bcl-xL expression. Control HMECs were serum starved for 24 hours. HMECs serum-starved were stimulated with 10 μg/mL MVs or RNase-treated MVs for 24 hours. (C) Incubation of HMECs with MVs induced expression of e-NOS; the eNOS activation was indicated by its phosphorylation at ser1177 (P-eNOS). (D) Organization of HMECs into capillary-like structures on Matrigel was inhibited by PI3K inhibitors wortmannin (0.1 μM) and LY294002 (10 μM) and by the NOS inhibitors L-NAME (10 μM) and L-NMMA (10 μM). D-NAME and D-NMMA were ineffective. Data are expressed as mean (± 1 SD) from 3 different experiments. Analysis of variance with Newmann-Keuls multicomparison test was performed; *P < .05 MVs, MV + D-NAME or MVs + D-NMMA versus vehicle; §P < .05 MVs + PI3K or NOS inhibitors versus MVs.
MVs induced Akt activation and expression of Bcl-xL and eNOS. Cell lysates (30 μg of protein) were immunoblotted with anti–P-Akt, -Akt, – Bcl-xL or -βactin Abs. (A) Densitometric analysis of P-Akt/Akt ratio. Data of P-Akt/Akt ratio are expressed as mean (± 1 SD) from 3 different experiments. (B) Representative immunoblots of P-Akt, Akt, and Bcl-xL expression. Control HMECs were serum starved for 24 hours. HMECs serum-starved were stimulated with 10 μg/mL MVs or RNase-treated MVs for 24 hours. (C) Incubation of HMECs with MVs induced expression of e-NOS; the eNOS activation was indicated by its phosphorylation at ser1177 (P-eNOS). (D) Organization of HMECs into capillary-like structures on Matrigel was inhibited by PI3K inhibitors wortmannin (0.1 μM) and LY294002 (10 μM) and by the NOS inhibitors L-NAME (10 μM) and L-NMMA (10 μM). D-NAME and D-NMMA were ineffective. Data are expressed as mean (± 1 SD) from 3 different experiments. Analysis of variance with Newmann-Keuls multicomparison test was performed; *P < .05 MVs, MV + D-NAME or MVs + D-NMMA versus vehicle; §P < .05 MVs + PI3K or NOS inhibitors versus MVs.
Discussion
In the present study we demonstrated that MVs derived from EPCs are able to trigger angiogenesis both in vitro and in vivo by a horizontal transfer of mRNA to human micro vascular and macrovascular endothelial cells.
MVs are released by various cell types such as circulating blood cells and cells of the vessel wall during cell activation by agonists and physical or chemical stress.32,33 MVs represent a heterogeneous population, differing in cellular origin, number, size, antigenic composition, and functional properties.34,35 In vivo, the majority of MVs is derived from platelets,36 whereas only few circulating MVs are derived from other blood cells and from endothelial cells. At their surface, MVs express the characteristic antigens of the cell from which they originated and in addition can expose activation markers and carry other membrane and cytoplasmic constituents.35 MVs may interact with target cells by surface-expressed ligands, transfer surface receptors, deliver proteins, mRNA, and bioactive lipids. Moreover, they may serve as a vehicle for the transfer of infectious particles (HIV, prions) and perhaps deliver intact organelles, representing important modulators of cell-to-cell communication.19
Ratajczak et al18 recently showed that MVs derived from embryonic stem cells may reprogram hematopoietic progenitors. This study clearly demonstrated that MVs may contribute to the developmental program of target cells either by a horizontal transfer of mRNA or by protein delivery.
In the present study we tested the hypothesis that MVs derived from EPCs may activate an angiogenic program in mature quiescent endothelial cells. Data derived from EPC-based cell therapy, despite the beneficial effects, did not clarify whether these cells directly contributed to revascularization of ischemic tissue or rather favored neoangiogenesis by a paracrine mechanism.37,38 Also data relative to the role of EPCs in tumor angiogenesis remain controversial.39 It has been suggested that transdifferentiation or plasticity of stem cells may at least in part depend on horizontal transfer of mRNA/proteins from the damaged tissue.18 Conversely, MV-mediated transfer of mRNA/proteins derived from stem cells may induce dedifferentiation of mature cells, triggering a proliferative program that may contribute to the repair of tissue injury. Previous studies showed that MVs derived from platelets stimulate in vitro proliferation, migration, and tube formation in endothelial cells mediated by their lipid components.40 Moreover, it has been shown that metalloproteinases harbored by endothelial MVs regulate the proteolytic activity on matrix required to elicit angiogenesis.41
In the present study we demonstrate that MVs derived from EPCs may induce proliferation, apoptosis resistance, and in vitro organization in capillary-like structures in HMECs and HUVECs. It has been previously reported that MVs may express surface molecules characteristic of originating cells35 and adhesion molecules and receptors.8,9,42 We found that EPC-derived MVs expressed several adhesion molecules such as ICAM-1, α4 integrin, CD44, and CD29 known to be present on the surface of EPCs.21 Some of these molecules, namely α4 integrin and CD29 (β1 integrin), were found to be instrumental in MV internalization into HMECs and HUVECs because blocking Abs prevented MV incorporation. Moreover, we found that the internalization of MVs was required for their biologic activity, but the interaction with the adhesion molecules was not the main mechanism of cell activation because the RNase treatment almost completely abrogated the MV-induced activation of the angiogenic program. The dependency on mRNA transfer was particularly evident for the MV-induced antiapoptotic effect and stimulation of endothelial cell organization in capillary-like structures. These results suggest that adhesion molecules did not directly mediate stimulation of endothelial cells, but that they were critical for MV internalization. Moreover, the primary role of mRNA horizontal transfer in the activation of angiogenesis was shown by experiments of endothelial cell stimulation with MV-mRNA extracts vehiculated by lipofectamine. These results are consistent with previous reports indicating that MVs may deliver mRNA to target cells18 and that the transfer of small amounts of exogenous mRNA can indeed modulate the cellular behavior.43 To evaluate whether MVs derived from EPCs were enriched for mRNA encoding specific proteins, we performed a microarray analysis. We found a total of 298 transcripts, 183 of which associated to RefSeq identifiers and remaining Unigene EST, suggesting that particles did not contain a random sample of cellular mRNA, but rather a specific subset. In particular, we found transcripts associated with the PI3K/AKT signaling pathway and with eNOS, known to be involved in the angiogenic and antiapoptotic program.44,45 Interestingly, MVs carried also the gene POLR2B encoding the polymerase responsible for synthesizing mRNA in eukaryotes. This subunit, in combination with at least 2 other polymerase subunits, forms a structure within the polymerase that maintains contact in the active site of the enzyme between the DNA template and the newly synthesized RNA.46 We also demonstrated that MVs may trigger the activation of PI3K/Akt signaling pathway and eNOS in target endothelial cells by enhancing the protein expression and phosphorylation of Akt and eNOS. Moreover, MVs induced the expression of the antiapoptotic protein Bcl-xL in target endothelial cells. Because mRNA encoding for these molecules was present in MVs and the expression of the corresponding proteins was abrogated by pretreatment of MVs with RNase, it can be suggested that mRNA derived from MVs was instrumental in protein transduction. Blockade of PI3K/Akt signaling pathway and of eNOS prevented MV-induced in vitro angiogenesis, suggesting their critical role in the activation of endothelial angiogenic program by MVs.
It is not clear at present how such MV-mediated mRNA transfer is involved in in vivo cell-to-cell communication. The present study indicates that MVs may also trigger angiogenesis in vivo by mRNA transfer as the angiogenic activity is abrogated by MV pretreatment with RNase.
The study of Ratajczak,18 as well as the present study, open new research perspectives on the use of MVs to transfer RNA-based information from stem cells/precursors to target differentiated cells. In particular, the results of the present study indicate that MVs derived from EPCs may activate an angiogenic program in quiescent endothelial cells.
The observation that MVs derived from stem cells/precursors are specifically enriched for certain proteins and transcription factors suggests the existence of a cellular mechanism for selective compartmentalization of such factors into MVs that may be exploited in regenerative medicine.
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC), by the Italian Ministry of University and Research (MIUR) COFIN and ex60%, by the Regione Piemonte integrated project A47, by the Italian Ministry of Health (Ricerca Finalizzata 02), and by Progetto S. Paolo (G.C.).
Authorship
Contribution: M.C.D., B.B., and G.C. designed the research; M.C.D., V.C., C.T., L.B., and S.B. performed the research; R.C. and M.L.I. performed the gene array analysis; and M.C.D., V.C., B.B., and G.C. analyzed the data and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: G. Camussi, Dipartimento di Medicina Interna, Ospedale Maggiore S. Giovanni Battista, Corso Dogliotti 14, 10126, Torino, Italy; e-mail:giovanni.camussi@unito.it.
