

Abstract
The induction of CD11b+Gr-1+ myeloid-derived suppressor cells (MDSCs) is an important immune-evading mechanism used by tumors. However, the exact nature and function of MDSCs remain elusive, especially because they constitute a heterogeneous population that has not yet been clearly defined. Here, we identified 2 distinct MDSC subfractions with clear morphologic, molecular, and functional differences. These fractions consisted of either mononuclear cells (MO-MDSCs), resembling inflammatory monocytes, or low-density polymorphonuclear cells (PMN-MDSCs), akin to immature neutrophils. Interestingly, both MO-MDSCs and PMN-MDSCs suppressed antigen-specific T-cell responses, albeit using distinct effector molecules and signaling pathways. Blocking IFN-γ or disrupting STAT1 partially impaired suppression by MO-MDSCs, for which nitric oxide (NO) was one of the mediators. In contrast, while IFN-γ was strictly required for the suppressor function of PMN-MDSCs, this did not rely on STAT1 signaling or NO production. Finally, MO-MDSCs were shown to be potential precursors of highly antiproliferative NO-producing mature macrophages. However, distinct tumors differentially regulated this inherent MO-MDSC differentiation program, indicating that this phenomenon was tumor driven. Overall, our data refine tumor-induced MDSC functions by uncovering mechanistically distinct MDSC subpopulations, potentially relevant for MDSC-targeted therapies.
Introduction
Tumors use a wide array of immunosuppressive strategies by which they blunt immune responses and limit the effect of immunotherapy.1 In this regard, tumors are found to affect myelopoiesis and induce the expansion of myeloid cells with immunosuppressive activity, in both animal models and human patients.2-10 These cells are referred to as myeloid-derived suppressor cells (MDSCs), which are in mice typically identified by the coexpression of the CD11b and Gr-1 surface markers.11 Tumor-induced CD11b+Gr-1+ cells accumulate in the bone marrow, spleen, and blood, and are considered to be a heterogeneous population consisting of different myeloid cell types in various maturation states. Despite this notion, these cells are typically studied as a bulk population and only a few reports have addressed a further refinement of this population.3,8,12
It has been suggested that different tumor-derived factors, such as granulocyte macrophage–colony-stimulating factor (GM-CSF), vascular endothelial growth factor (VEGF), IL-1β, and prostaglandins, can mobilize MDSC recruitment from bone marrow hematopoietic precursors and induce the immunosuppressive phenotype.13-18 While this immunosuppressive phenotype is a trademark of MDSCs, there is no clear consensus on their suppressive mechanism. Nevertheless, the l-arginine metabolism often seems to play a central role in the immune-suppressive activity of MDSCs. l-arginine can be metabolized by inducible nitric oxide synthase (iNOS or Nos2), generating citrulline and nitric oxide (NO), or can be converted into urea and l-ornithine by arginase.19 MDSCs expressing arginase-1 reduce the availability of l-arginine, which can result in the loss of CD3ζ expression and impaired T-cell function.7 Others have shown that production of NO by iNOS-expressing MDSCs is sufficient for blocking T-cell responses.8 Furthermore, it has been suggested that the simultaneous expression of both enzymes can result in superoxide production, ultimately giving rise to T cell–suppressive reactive nitrogen-oxide species (RNOSs) such as peroxynitrite.20 Other studies have shown that MDSCs can produce high amounts of the T cell–suppressive reactive oxygen species (ROS) H2O2 after antigen-specific interaction with CD8+ T cells.5 Whether these different suppressor functions of MDSCs can be attributed to distinct MDSC subpopulations is unknown.
In this study, we defined the phenotype of discrete MDSC subfractions from 2 genetically unrelated T-cell lymphoma models and assessed their suppressive potency and mechanism toward antigen-driven or polyclonal T-cell stimulation, using different in vitro stimulation protocols. Interestingly, although both mononuclear and polymorphonuclear MDSCs suppress antigen-specific T-cell responses through an at least partially IFN-γ–regulated mechanism, they use distinct signaling pathways and effector molecules. Moreover, we show that suppression of polyclonal T-cell responses depends on the ability of the mononuclear MDSC fraction to efficiently differentiate toward suppressive macrophages. Of importance, the latter capacity is differentially regulated by different tumors.
Methods
Mice, cell lines, and tumor growth
Six- to 9-week-old female AKR (H-2k) and C57BL/6 (H-2b) mice were purchased from Harlan (Horst, The Netherlands) and (AKR × C57BL/6)F1 mice were bred in our animal facilities. OT-1 mice were a gift of Dr Muriel Moser (ULB, Brussels, Belgium). C57BL/6 β2-microglobulin−/−, STAT1−/−, and iNOS−/− mice were provided by Dr Georges Leclercq (UZGent, Ghent, Belgium), Dr Chantal Mathieu (KUL, Leuven, Belgium), and Dr Anje Cauwels (UGent, Ghent, Belgium), respectively.
The BW-Sp3 cell line was obtained from the original BW5147 T-cell lymphoma, and EG7 is a chicken ovalbumin–transfected EL-4 thymoma, as described earlier.21,22 Cancer cells were maintained in RPMI1640 medium, supplemented with 10% heat-inactivated FCS, 0.03% l-glutamine, 100 mg/mL streptomycin, and 100 mg/mL penicillin (Invitrogen, Frederick, MD). For splenocyte cultures, this medium was further supplemented with 1 mM nonessential amino acids, 1 mM sodium pyruvate (Invitrogen), and 0.02 mM 2-ME.
Mice were injected subcutaneously in the flank with 3 × 106 BW-Sp3 or EG7 cells and were used when average tumor diameters reached 15 to 20 mm.
Fluorescence-activated cell sorting staining and flow cytometry
Information on commercial antibodies can be found in Table 1. 7/4 was a gift of Dr Gordon Brown (Sir William Dunn School of Pathology, Oxford, United Kingdom). Nonlabeled anti-CCR2 (MC-21) was a gift of Dr Matthias Mack (University of Regensburg, Regensburg, Germany). Fractalkine-Fc and antihumanFc/Cy5, to assess the expression of CX3CR1, were provided by Dr Steffen Jung (Weizmann Institute of Science, Rehovot, Israel). Nonlabeled anti–SIGN-R1 (ER-TR9) and anti-MGL1/2 (ER-MP23) were provided by Dr Pieter Leenen (Erasmus MC, Rotterdam, The Netherlands).
List of commercial antibodies used
| Markers | Clone | Manufacturer |
|---|---|---|
| CD11b/PE/FITC/APC | M1/70 | BD Biosciences (San Jose, CA) |
| Gr-1/FITC | RB6-8C5 | BD Biosciences |
| Ly6G/FITC/PE | 1A8 | BD Biosciences |
| CD34/bio | RAM34 | BD Biosciences |
| CD117/bio | 2B8 | BD Biosciences |
| Sca-1/bio | D7 | BD Biosciences |
| F4/80/PE | CI:A3-1 | Serotec (Raleigh, NC) |
| CD115/PE | AFS98 | eBioscience (San Diego, CA) |
| CD62L/PE | SK11 | BD Biosciences |
| Ly6C /FITC | AL21 | BD Biosciences |
| Tie-2/purified | 1E11DH | Millipore (Billerica, MA) |
| CD49d/PE | 9C10(MFR4. B) | BD Biosciences |
| CD11a/purified | 2D7 | BD Biosciences |
| CD2/purified | RM2–5 | BD Biosciences |
| CD31/FITC | MEC13.3 | BD Biosciences |
| CD162/PE | 2PH1 | BD Biosciences |
| CD54/bio | 3E2 | BD Biosciences |
| CD43/PE | S7 | BD Biosciences |
| CD44/PE | IM7 | BD Biosciences |
| CD14/PE | RMC5-3 | BD Biosciences |
| CD204/RPE | 2F8 | Serotec |
| MAC-2/FITC | M3/38 | Cedarlane (Burlington, ON) |
| CD16/32/PE | 2.4G2 | BD Biosciences |
| CD23/PE | B3B4 | BD Biosciences |
| CD21/35/FITC | 7G6 | BD Biosciences |
| CD13/PE | R3-242 | BD Biosciences |
| CD40/FITC | 3/23 | BD Biosciences |
| CD86/FITC | GL-1 | BD Biosciences |
| CD1d/PE | 1B1 | BD Biosciences |
| H-2Kb/FITC | AF6-88.5 | BD Biosciences |
| H-2Dk/FITC | 15-5-5 | BD Biosciences |
| IA/IE/PE | M5/114.15.2 | BD Biosciences |
| CD80/FITC | 16-10A1 | BD Biosciences |
| B7H1/PE | MIH5 | eBioscience |
| B7DC/PE | TY25 | eBioscience |
| B7H4/PE | eBioMIH29 | eBioscience |
| CD124 (IL4Rα)/PE | mIL4RM1 | BD Biosciences |
| CD71/FITC | R17217 | eBioscience |
| CCR3/FITC | 83101 | R&D Systems (Minneapolis, MN) |
| Markers | Clone | Manufacturer |
|---|---|---|
| CD11b/PE/FITC/APC | M1/70 | BD Biosciences (San Jose, CA) |
| Gr-1/FITC | RB6-8C5 | BD Biosciences |
| Ly6G/FITC/PE | 1A8 | BD Biosciences |
| CD34/bio | RAM34 | BD Biosciences |
| CD117/bio | 2B8 | BD Biosciences |
| Sca-1/bio | D7 | BD Biosciences |
| F4/80/PE | CI:A3-1 | Serotec (Raleigh, NC) |
| CD115/PE | AFS98 | eBioscience (San Diego, CA) |
| CD62L/PE | SK11 | BD Biosciences |
| Ly6C /FITC | AL21 | BD Biosciences |
| Tie-2/purified | 1E11DH | Millipore (Billerica, MA) |
| CD49d/PE | 9C10(MFR4. B) | BD Biosciences |
| CD11a/purified | 2D7 | BD Biosciences |
| CD2/purified | RM2–5 | BD Biosciences |
| CD31/FITC | MEC13.3 | BD Biosciences |
| CD162/PE | 2PH1 | BD Biosciences |
| CD54/bio | 3E2 | BD Biosciences |
| CD43/PE | S7 | BD Biosciences |
| CD44/PE | IM7 | BD Biosciences |
| CD14/PE | RMC5-3 | BD Biosciences |
| CD204/RPE | 2F8 | Serotec |
| MAC-2/FITC | M3/38 | Cedarlane (Burlington, ON) |
| CD16/32/PE | 2.4G2 | BD Biosciences |
| CD23/PE | B3B4 | BD Biosciences |
| CD21/35/FITC | 7G6 | BD Biosciences |
| CD13/PE | R3-242 | BD Biosciences |
| CD40/FITC | 3/23 | BD Biosciences |
| CD86/FITC | GL-1 | BD Biosciences |
| CD1d/PE | 1B1 | BD Biosciences |
| H-2Kb/FITC | AF6-88.5 | BD Biosciences |
| H-2Dk/FITC | 15-5-5 | BD Biosciences |
| IA/IE/PE | M5/114.15.2 | BD Biosciences |
| CD80/FITC | 16-10A1 | BD Biosciences |
| B7H1/PE | MIH5 | eBioscience |
| B7DC/PE | TY25 | eBioscience |
| B7H4/PE | eBioMIH29 | eBioscience |
| CD124 (IL4Rα)/PE | mIL4RM1 | BD Biosciences |
| CD71/FITC | R17217 | eBioscience |
| CCR3/FITC | 83101 | R&D Systems (Minneapolis, MN) |
Intracellular Foxp3 staining was performed following the manufacturer's instructions (eBioscience, San Diego, CA). Tumor dissociation was performed as described earlier.12
Fluorescence-activated cell sorting (FACS) data were acquired using a FACSVantage SE flow cytometer or FACS Canto 2 (BD Biosciences).
Isolation of MDSCs and MDSC subfractions
To purify tumor-induced CD11b+Gr-1+ cells, erythrocyte-depleted splenocytes were first depleted of CD11b+Gr-1− cells (mostly being CD19+ or CD11c+, markers that were furthermore not expressed on CD11b+Gr-1+ cells) via magnetic selection using anti-CD19 and anti-CD11c microbeads and LD columns following the manufacturer's instructions (Miltenyi Biotec, Auburn, CA). In the remaining population, CD11b+ cells were positively selected via anti-CD11b microbeads using LS columns (Miltenyi Biotec), providing purified total MDSCs. To purify Ly6G+ MDSCs, CD19/CD11c-depleted splenocytes from tumor-bearing mice were stained with biotin-coupled anti-Ly6G, followed by positive magnetic selection using antibiotin microbeads, following the manufacturer's instructions (anti-Ly6G microbead kit; Miltenyi Biotec). To further purify Ly6G− MDSCs from the same spleen, remaining cells were passed over an LD depletion column to make sure all Ly6G+ cells were eliminated, followed by a positive selection using anti-CD11b microbeads. The purity of the total MDSC population or the MDSC subfractions was typically higher than 90%.
Morphologic analysis
Cytospins were obtained by spinning down cells on precoated microscope slides (Thermo Shandon, Pittsburgh, PA), fixing with methanol and staining with May-Grünwald and Giemsa dye for 5 and 15 minutes, respectively. Images were obtained using the Nikon Eclipse E600 microscope (Nikon, Melville, NY) with Nikon Plan lenses at 40×/0.65 magnification and without immersion oil. Pictures were taken using a Vision Technologies Basler camera (Vision Technologies, Glen Burnie, MD), acquired with Lucia Image Archive Plus Version 4.2 (Laboratory Imaging, Prague, Czech Republic) and processed with Adobe Photoshop CS1 (Adobe Systems, San Jose, CA).
Suppression of antigen-specific T-cell proliferation
Purified MDSCs were added in various amounts to 2 × 105 naive OT-1 splenocytes, in flat-bottom 96-well plates. These cocultures were promptly stimulated with 250 μg/mL chicken ovalbumin protein (Sigma-Aldrich, St Louis, MO); 24 hours later 1 μCi (0.037 MBq) 3H-thymidine (Amersham, Arlington Heights, IL) was added and cells were allowed to proliferate for another 18 hours before incorporated radioactivity was measured (= short-term culture protocol, 24 + 18 = 42 hours total). Anti–IFN-γ (10 μg/mL, clone F3; provided by Dr Hubertine Heremans, Rega Institute, Leuven, Belgium), L-NMMA (0.5 mM; Sigma-Aldrich), nor-NOHA (0.5 mM; Calbiochem, San Diego, CA), SC-791 (100 nM; Calbiochem), catalase (1000 U/mL; Sigma-Aldrich), and superoxide dismutase (SOD, 200 U/mL; Sigma-Aldrich) were added from the beginning of the culture. Percentage suppression of proliferation without inhibitor is calculated as

Relative percentage suppression of proliferation with inhibitor is calculated as

Relative percentage suppression is calculated as
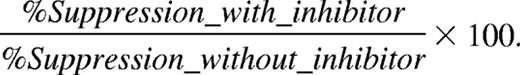
Suppression of polyclonal T-cell proliferation
Purified MDSCs were added in various amounts to 2 × 105 naive AKR, C57BL/6, or (AKR × C57BL/6)F1 splenocytes, in flat-bottom 96-well plates. These cocultures were promptly stimulated with 1 μg/mL anti-CD3 (or alternatively 2.5 μg/mL concanavalin A); 24 hours later 3H-thymidine was added and cells were allowed to proliferate for another 18 hours before measurement (= short-term culture protocol). Alternatively, MDSCs and splenocytes were cocultured for 48 hours, at which time point anti-CD3 was added. Again, 3H-thymidine was added 24 hours later and cells were allowed to proliferate for another 18 hours before measurement (= long-term culture protocol). Anti–IFN-γ, L-NMMA, and nor-NOHA were added simultaneously with anti-CD3.
Quantification of NO2−
Nitrite quantification was assayed by a standard Greiss reaction, as described previously.9
Statistical analysis
Statistical significance was determined by the unpaired Student t test or the paired Student t test (only for Figure 4C), using GraphPad Prism 4.0 software (GraphPad Software, San Diego, CA).
Results
CD11b+Gr-1+ MDSCs from tumor-bearing mice consist of a mononuclear and polymorphonuclear fraction
To refine MDSC heterogeneity, we used 2 unrelated mouse T-cell lymphoma models—EG7 and BW-Sp3—derived from the C57BL/6 and AKR strain, respectively. As previously reported,6,9 subcutaneous inoculation of these cancer cells resulted in a profound expansion of splenic CD11b+Gr-1+ MDSCs within a few weeks of tumor progression (Figure 1A). Interestingly, in both models, purified tumor-induced CD11b+Gr-1+ cells consisted of 2 major subfractions based on differential Ly6G expression (Figure 1Bi). The Ly6G− and Ly6G+ fractions each represented approximately half of the total CD11b+Gr-1+ population (EG7: 48% ± 4% Ly6G− and 52% ± 4% Ly6G+; BW-Sp3: 41% ± 2% Ly6G− and 59% ± 2% Ly6G+), indicating that both MDSC subpopulations were equally driven to expand under the influence of tumor growth. To further investigate the characteristics of these MDSC subfractions, they were purified to near uniformity. While purified Ly6G+ MDSCs displayed a high SSC profile (Figure 1Bii) in accordance with their polymorphonuclear morphology (Figure 1C; these cells will be termed PMN-MDSCs), Ly6G− MDSCs were mononuclear (Figure 1C; these cells will be termed MO-MDSCs) and had on average a lower SSC (Figure 1Biii). Moreover, the observed heterogeneity in Gr-1 expression of CD11b+Gr-1+ cells results from the fact that the Ly6G+ fraction almost uniformly corresponded to Gr-1 high expressers, while Ly6G− cells had on average a lower Gr-1 expression (Figure 1Bii,iii).

Splenic CD11b+Gr-1+ MDSCs from tumor-bearing mice consist of a Ly6G+SSChi polymorphonuclear and a Ly6G−SSClo mononuclear fraction. (A) Splenocytes were collected from BW-Sp3 tumor–bearing AKR mice and EG7 tumor–bearing C57BL/6 mice, or their respective naive counterparts and were stained with PE-labeled anti-CD11b and FITC-labeled anti–Gr-1 mAbs. The percentage of CD11b+Gr-1+ cells is indicated. (Bi) CD11b+Gr-1+ cells from tumor-bearing mice were purified as described in “Methods.” These cells were stained with PE-conjugated anti-CD11b and either FITC-conjugated anti–Gr-1 or anti-Ly6G. Because the anti–Gr-1 mAb RB6–8C5 recognizes both the Ly6G and Ly6C molecules, costaining with the anti-Ly6G mAb 1A8 was not possible due to overlapping epitopes, as previously reported.23 Gate R1 represents MDSCs with an intermediate to high Gr-1 expression level, while gate R2 represents the highest Gr-1 expressers. Gate R3 represents Ly6G− MDSC, and gate R4 represents Ly6G+ cells. (Bii,iii) Ly6G+ and Ly6G− MDSCs were purified as described in “Methods” and were stained with the indicated mAbs. Percentages represent the purity of isolated populations. In addition, a dot plot representing the FSC/SSC profile of the purified cells is shown. Median fluorescence intensity (MFI) of the corresponding Gr-1 expression levels and SSC profiles is given. (C) Purified MDSC fractions were subjected to cytospin and May-Grünwald-Giemsa staining. Pictures from BW-Sp3 and EG7 MDSC subfractions are shown (×40 magnification).
Splenic CD11b+Gr-1+ MDSCs from tumor-bearing mice consist of a Ly6G+SSChi polymorphonuclear and a Ly6G−SSClo mononuclear fraction. (A) Splenocytes were collected from BW-Sp3 tumor–bearing AKR mice and EG7 tumor–bearing C57BL/6 mice, or their respective naive counterparts and were stained with PE-labeled anti-CD11b and FITC-labeled anti–Gr-1 mAbs. The percentage of CD11b+Gr-1+ cells is indicated. (Bi) CD11b+Gr-1+ cells from tumor-bearing mice were purified as described in “Methods.” These cells were stained with PE-conjugated anti-CD11b and either FITC-conjugated anti–Gr-1 or anti-Ly6G. Because the anti–Gr-1 mAb RB6–8C5 recognizes both the Ly6G and Ly6C molecules, costaining with the anti-Ly6G mAb 1A8 was not possible due to overlapping epitopes, as previously reported.23 Gate R1 represents MDSCs with an intermediate to high Gr-1 expression level, while gate R2 represents the highest Gr-1 expressers. Gate R3 represents Ly6G− MDSC, and gate R4 represents Ly6G+ cells. (Bii,iii) Ly6G+ and Ly6G− MDSCs were purified as described in “Methods” and were stained with the indicated mAbs. Percentages represent the purity of isolated populations. In addition, a dot plot representing the FSC/SSC profile of the purified cells is shown. Median fluorescence intensity (MFI) of the corresponding Gr-1 expression levels and SSC profiles is given. (C) Purified MDSC fractions were subjected to cytospin and May-Grünwald-Giemsa staining. Pictures from BW-Sp3 and EG7 MDSC subfractions are shown (×40 magnification).
To gain further insight in the maturation state, lineage commitment, and potential functions of these MDSC subpopulations, their surface marker expression was extensively studied (Table 2). MDSCs did not express the stem cell/progenitor markers CD117 and CD34. However, Sca-1, which is present on both stem cells and on committed Lin+ cells in early stages of differentiation,24 was expressed solely on the MO-MDSCs from BW-Sp3 tumor–bearing mice, suggesting a difference in maturation state between the different MDSCs. Irrespective of their level of maturation, the majority of Ly6G− MO-MDSCs from both tumor models expressed the typical monocyte/macrophage markers F4/80 and CD115, indicating that these cells were committed to the monocyte lineage. More precisely, MO-MDSCs from EG7 and BW-Sp3 tumor–bearing mice resemble the “inflammatory” fraction of peripheral blood monocytes,25,26 based on the profile Ly6ChighCCR2+CX3CR1−/lowCD62L+. Tie2, a characteristic marker of proangiogenic monocytes in tumor-bearing mice,27 was absent from MO-MDSCs.
Surface marker expression of MO-MDSCs and PMN-MDSCs from EG7 and BW-Sp3 tumor–bearing mice
| MO-MDSC | PMN-MDSC | |||
|---|---|---|---|---|
| EG7 | Sp3 | EG7 | Sp3 | |
| Myeloid maturation markers | ||||
| CD34 | − | − | − | − |
| CD117 (c-kit) | − | − | − | − |
| Sca-1 (Ly6A/E)* | − | + | − | − |
| Myeloid lineage markers | ||||
| F4/80† | ++ | ++ | +/− | − |
| CD115 (M-CSFR)† | + | + | − | − |
| 7/4‡ | +++ | +++ | ++ +/− | ++ +/− |
| Monocyte/PMN subset markers | ||||
| CCR2‡ | ++ | ++ | + | + |
| CX3CR1 | − | − | − | − |
| CD62L | +++ | +++ | +++ | +++ |
| Ly6C‡ | +++ | +++ | ++ +/− | ++ +/− |
| Tie-2 | − | − | − | − |
| VLA-4 (CD49d)‡ | +++ | +++ | + | + |
| Adhesion molecules | ||||
| CD11a (LFA-1) | ++ | ++ | ++ | ++ |
| CD2 (LFA-2)† | + | + | − | − |
| CD31 (PECAM-1)† | + | + | − | − |
| CD162 (PSGL-1) | +++ | +++ | +++ | +++ |
| CD54 (ICAM-1)‡ | +++ | +++ | + | + |
| CD43 | +++ | +++ | +++ | +++ |
| CD44 | +++ | +++ | +++ | +++ |
| Pattern recognition receptors | ||||
| CD14 | − | − | − | − |
| CD204 (SR-A) | − | − | − | − |
| SIGN-R1 | − | − | − | − |
| MAC-2 (Galectin-3)‡ | ++ | ++ | + | + |
| Fc/complement receptors | ||||
| CD16/32 (FcγRIII/II) | + | + | + | + |
| CD23 (FcϵRII) | − | − | − | − |
| CD21/35 (CR2/CR1) | − | − | − | − |
| Antigen processing/presentation | ||||
| CD13 | − | − | − | − |
| CD40 | − | − | − | − |
| CD86 (B7–2) | − | − | − | − |
| CD1d† | + | + | − | − |
| MHC I | +++ | +++ | +++ | +++ |
| MHC II | − | − | − | − |
| Potential T cell–suppressive markers | ||||
| CD80 (B7–1) | ++ | ++ | ++ | ++ |
| B7H1 (PD-L1) | + | + | + | +/− |
| B7DC (PD-L2)* | +/− | − | +/− | − |
| B7H4 | − | − | − | − |
| MGL1/2† | + | + | − | − |
| Miscellaneous | ||||
| IL4Rα | + | + | + | + |
| CD71 (transferrin receptor)† | + | + | − | − |
| MO-MDSC | PMN-MDSC | |||
|---|---|---|---|---|
| EG7 | Sp3 | EG7 | Sp3 | |
| Myeloid maturation markers | ||||
| CD34 | − | − | − | − |
| CD117 (c-kit) | − | − | − | − |
| Sca-1 (Ly6A/E)* | − | + | − | − |
| Myeloid lineage markers | ||||
| F4/80† | ++ | ++ | +/− | − |
| CD115 (M-CSFR)† | + | + | − | − |
| 7/4‡ | +++ | +++ | ++ +/− | ++ +/− |
| Monocyte/PMN subset markers | ||||
| CCR2‡ | ++ | ++ | + | + |
| CX3CR1 | − | − | − | − |
| CD62L | +++ | +++ | +++ | +++ |
| Ly6C‡ | +++ | +++ | ++ +/− | ++ +/− |
| Tie-2 | − | − | − | − |
| VLA-4 (CD49d)‡ | +++ | +++ | + | + |
| Adhesion molecules | ||||
| CD11a (LFA-1) | ++ | ++ | ++ | ++ |
| CD2 (LFA-2)† | + | + | − | − |
| CD31 (PECAM-1)† | + | + | − | − |
| CD162 (PSGL-1) | +++ | +++ | +++ | +++ |
| CD54 (ICAM-1)‡ | +++ | +++ | + | + |
| CD43 | +++ | +++ | +++ | +++ |
| CD44 | +++ | +++ | +++ | +++ |
| Pattern recognition receptors | ||||
| CD14 | − | − | − | − |
| CD204 (SR-A) | − | − | − | − |
| SIGN-R1 | − | − | − | − |
| MAC-2 (Galectin-3)‡ | ++ | ++ | + | + |
| Fc/complement receptors | ||||
| CD16/32 (FcγRIII/II) | + | + | + | + |
| CD23 (FcϵRII) | − | − | − | − |
| CD21/35 (CR2/CR1) | − | − | − | − |
| Antigen processing/presentation | ||||
| CD13 | − | − | − | − |
| CD40 | − | − | − | − |
| CD86 (B7–2) | − | − | − | − |
| CD1d† | + | + | − | − |
| MHC I | +++ | +++ | +++ | +++ |
| MHC II | − | − | − | − |
| Potential T cell–suppressive markers | ||||
| CD80 (B7–1) | ++ | ++ | ++ | ++ |
| B7H1 (PD-L1) | + | + | + | +/− |
| B7DC (PD-L2)* | +/− | − | +/− | − |
| B7H4 | − | − | − | − |
| MGL1/2† | + | + | − | − |
| Miscellaneous | ||||
| IL4Rα | + | + | + | + |
| CD71 (transferrin receptor)† | + | + | − | − |
Expression of the indicated markers was evaluated on gated CD11b+Ly6C+Ly6G−(Gr-1+) MO-MDSC and CD11b+Ly6C+Ly6G+(Gr-1+) PMN-MDSC subfractions, relative to isotype-matched controls.
Markers differing between the 2 tumor models.
Markers expressed on MO-MDSCs but not on PMN-MDSCs.
Markers differentially expressed between MO- and PMN-MDSCs.
Expression of the typical neutrophil marker Ly6G,28 in combination with their polymorphonuclear morphology, suggests that PMN-MDSCs are committed to the neutrophil lineage. However, in contrast to mature peripheral blood neutrophils, PMN-MDSCs were found to copurify with mononuclear cells when run over a density gradient (data not shown), indicating their low-density phenotype.
Having established the lineage of MDSC subfractions by morphology and lineage marker expression, we next screened for additional surface markers that, independently of the tumor model, are able to discriminate between MO-MDSCs and PMN-MDSCs. CD1d, CD2, CD31, CD71, and MGL1/2 were detected on the MO-MDSC fraction in both models, while these molecules were absent from the PMN-MDSCs (Table 2). Other markers, such as VLA-4, CD54, CCR2, 7/4, and Mac-2 (galectin-3), were detected on both MDSC fractions, but reached higher levels on MO-MDSCs. Remarkably, the IL-4Rα chain, reported earlier as a marker for tumor-induced MDSCs,3 was equally expressed on MO-MDSCs and PMN-MDSCs.
Together, these results show that MDSCs can be clearly subdivided into 2 distinct populations. Furthermore, these subfractions were observed in independent tumor models, with an almost identical phenotype, suggesting that this is a general feature of tumor-induced MDSCs.
The phenotype and cellular composition of MO-MDSCs changes during the course of tumor growth, while that of PMN-MDSCs remains static
To determine the kinetics of MO- and PMN-MDSC induction, the percentage of these cells compared with the total splenocyte population was measured at various time points after tumor injection. This showed that MO- and PMN-MDSCs were induced with similar kinetics (Figure 2A).

Tumor-induced MO-MDSCs phenotypically differ from their naive counterparts. (A) Values represent percentages of CD11b+Ly6C+Ly6G+ (PMN-MDSCs) and CD11b+Ly6C+Ly6G− (MO-MDSCs) splenocytes, measured for naive mice or at various time points after tumor inoculation. For EG7 this was at 7, 14, 21, and 28 days of tumor growth. For BW-Sp3, which is a regressor/progressor model,12 early pro-gressors represent progressing tumors with a mean diameter of 10 plus or minus 2 mm, while late pro-gressors had a tumor diameter of 20 plus or minus 3 mm. (B) FSC/SSC plots are shown for gated CD11c−CD11b+Ly6C+Ly6G− splenocytes from naive mice (C57BL/6 and AKR) or tumor-bearing mice (EG7 and BW-Sp3). Gate R1 represents SSChighCD11b+F4/80+Ly6CintCCR3+ eosinophils, while cells in gate R2 are SSClowCD11b+F4/80+Ly6ChighCCR3− monocytes. Histogram plots represent expression of the indicated markers on naive gated (R1 or R2) cells compared with isotype controls (tinted). Histogram plots for R1- or R2-gated cells from tumor-bearing mice were identical (data not shown). (C) Histogram plots represent the expression of the indicated markers on gated (R2) CD11b+Ly6C+F4/80+CCR3− monocytic splenocytes from naive or tumor-bearing mice compared with isotype controls (tinted).
Tumor-induced MO-MDSCs phenotypically differ from their naive counterparts. (A) Values represent percentages of CD11b+Ly6C+Ly6G+ (PMN-MDSCs) and CD11b+Ly6C+Ly6G− (MO-MDSCs) splenocytes, measured for naive mice or at various time points after tumor inoculation. For EG7 this was at 7, 14, 21, and 28 days of tumor growth. For BW-Sp3, which is a regressor/progressor model,12 early pro-gressors represent progressing tumors with a mean diameter of 10 plus or minus 2 mm, while late pro-gressors had a tumor diameter of 20 plus or minus 3 mm. (B) FSC/SSC plots are shown for gated CD11c−CD11b+Ly6C+Ly6G− splenocytes from naive mice (C57BL/6 and AKR) or tumor-bearing mice (EG7 and BW-Sp3). Gate R1 represents SSChighCD11b+F4/80+Ly6CintCCR3+ eosinophils, while cells in gate R2 are SSClowCD11b+F4/80+Ly6ChighCCR3− monocytes. Histogram plots represent expression of the indicated markers on naive gated (R1 or R2) cells compared with isotype controls (tinted). Histogram plots for R1- or R2-gated cells from tumor-bearing mice were identical (data not shown). (C) Histogram plots represent the expression of the indicated markers on gated (R2) CD11b+Ly6C+F4/80+CCR3− monocytic splenocytes from naive or tumor-bearing mice compared with isotype controls (tinted).
Next, we wondered whether tumor-induced CD11b+Gr-1+ subsets phenotypically differed from their naive counterparts. In naive C57BL/6 or AKR mice, CD11b+Ly6C+Ly6G− (Gr-1+) cells consisted of 2 main subpopulations: approximately 30% to 40% of these cells were SSChighF4/80+Ly6CintCCR3+ cells (Figure 2B gate R1), which have been described as eosinophils,29,30 while the remainder were SSClowF4/80+Ly6ChighCCR3− monocytes (Figure 2B gate R2). However, as described earlier, in tumor-bearing hosts, the CD11b+Ly6C+Ly6G− MO-MDSC fraction consisted mainly of monocytes (typically > 90%) (Figure 2B), indicating that during tumor growth only CD11b+Ly6C+Ly6G− monocytes and CD11b+Ly6C+Ly6G+ neutrophilic PMN cells, but not eosinophils, were expanded. Subsequently, we wondered whether tumor-induced monocytes and Ly6G+ PMN cells phenotypically resembled their naive counterparts in terms of surface marker expression. No significant differences were found between CD11b+Ly6C+Ly6G+ PMN cells from naive and tumor-bearing hosts when considering the expression of the markers presented in Table 2 (data not shown). Naive and tumor-induced monocytes (gate R2) also showed a similar expression for the majority of these markers (data not shown). Interestingly however, in both tumor models, 3 markers were consistently found to be differentially expressed between naive and tumor-induced splenic monocytes (Figure 2C). These included the transferrin receptor CD71—which was detected only on tumor-induced monocytes—and CD80 and CD115, which reached a higher expression on tumor-induced compared with naive monocytes. As described earlier, Sca-1 was uniquely expressed on BW-Sp3–induced monocytes, and appeared to be absent from naive monocytes. Together these findings suggest that tumor-induced splenic MO-MDSCs are monocytes with a potentially unique activation and/or maturation status.
Finally, based on recent findings demonstrating a potential induction of Foxp3+ regulatory T cells (Tregs)8 by MDSCs, we investigated whether the induction of MDSCs relates to peripheral and intratumoral Treg levels in our tumor models. While the percentage of CD4+Foxp3+ Tregs was indeed slightly enhanced in the spleen of EG7 tumor-bearers, this did not reach statistical significance (Figure 3). In contrast, splenic Treg cell levels were significantly up-regulated in BW-Sp3 tumor–bearing mice, but a direct correlation with the number of splenic MDSCs was not unequivocal (Figure 3). Within tumors, the percentage of Tregs was invariably high throughout tumor growth and did not relate to the induction kinetics of MDSCs (Figure 3).

Kinetics of Foxp3+ regulatory T-cell induction in the spleen and tumor of EG7 or BW-Sp3 tumor-bearers. Intracellular Foxp3 staining was performed on single-cell suspensions of the spleen and tumor, measured at various time points after tumor inoculation, in parallel with the kinetic analysis of the induction of MDSCs (Figure 2A). Percentages indicate the number of CD4+Foxp3+ cells within the total CD4+ population. Each group consisted of at least 3 individual mice. *P < .05; ns indicates not significant.
Kinetics of Foxp3+ regulatory T-cell induction in the spleen and tumor of EG7 or BW-Sp3 tumor-bearers. Intracellular Foxp3 staining was performed on single-cell suspensions of the spleen and tumor, measured at various time points after tumor inoculation, in parallel with the kinetic analysis of the induction of MDSCs (Figure 2A). Percentages indicate the number of CD4+Foxp3+ cells within the total CD4+ population. Each group consisted of at least 3 individual mice. *P < .05; ns indicates not significant.
Both MO-MDSCs and PMN-MDSCs suppress antigen-specific T-cell responses
Next, we wondered to what extent each MDSC fraction contributes to the reported T cell–suppressive activity of CD11b+Gr-1+ cells. In first instance, purified total EG7 MDSCs were added in various amounts to TCR transgenic OT-1 splenocytes, which were stimulated in short-term cultures (42 hours) with either ovalbumin protein (OVA) or anti-CD3 (“Methods”). Remarkably, while EG7 MDSCs suppressed OVA-driven antigen-specific CD8+ T-cell proliferation, they did not affect anti-CD3–induced proliferation (Figure 4A). Interestingly, individually purified MO-MDSC and PMN-MDSC subfractions were both found to suppress OVA-driven T-cell proliferation dose dependently (Figure 4B). However, on average—when comparing the suppressive activity of both MDSC fractions over different experiments—MO-MDSCs were found to be more potent suppressors than PMN-MDSCs (Figure 4C). The observation that anti-CD3–induced T-cell proliferation was not affected in the presence of MDSCs suggested that MDSCs might use antigen-specific suppressive mechanisms. In this regard, some reports indicate that MDSC suppression requires antigen presentation through MHC I molecules.4,6 To investigate this mechanism, EG7 tumors were grown in β2-microglobulin–deficient mice, MO- and PMN-MDSCs were purified and added to OT-1 cultures together with OVA. These MDSC subfractions, deficient in MHC I expression,31 did not show any significant impairment in their suppressive activity (Figure 4D). Furthermore, OT-1 T-cell responses were also suppressed by allogeneic MO- and PMN-MDSCs originating from BW-Sp3 tumor–bearing AKR mice (Figure 4E). Hence, in this particular setting, an important role for antigen presentation by MDSCs in their suppressive activity can be excluded.

Both MDSC subfractions have the capacity to suppress antigen-driven T-cell responses. (A) OT-1 splenocytes were stimulated with either 1 μg/mL anti-CD3 or 250 μg/mL ovalbumin protein (OVA), in the presence of various amounts of purified total EG7 CD11b+Gr-1+ MDSCs, in a short-term culture (“Methods”). Graphs represent the average level of 3H-thymidine incorporation plus or minus SD, expressed as counts per minute (CPM), for each MDSC/splenocyte ratio. One representative experiment of 3 is shown. (B) Similar experiment as panel A, whereby OT-1 splenocytes were activated by OVA in the presence of various amounts of purified EG7 MO-MDSCs or PMN-MDSCs. One representative experiment of 8 is shown. (C) Percentage suppression induced by EG7 MO-MDSC and PMN-MDSC fractions at a 1:1 MDSC/splenocyte ratio, as observed in 8 individual experiments. (D) MO- and PMN-MDSCs were purified from EG7 tumor–bearing β2-microglobulin–deficient C57BL/6 mice and added to OT-1 cultures together with OVA. Percentage suppression represents the mean of 3 individual experiments. (E) OT-1 splenocytes were stimulated with OVA in the presence of BW-Sp3–induced AKR MO- or PMN-MDSCs (1:1 ratio) and percentage suppression was calculated. One representative experiment of 2 is shown. *P < .05; **P < .01.
Both MDSC subfractions have the capacity to suppress antigen-driven T-cell responses. (A) OT-1 splenocytes were stimulated with either 1 μg/mL anti-CD3 or 250 μg/mL ovalbumin protein (OVA), in the presence of various amounts of purified total EG7 CD11b+Gr-1+ MDSCs, in a short-term culture (“Methods”). Graphs represent the average level of 3H-thymidine incorporation plus or minus SD, expressed as counts per minute (CPM), for each MDSC/splenocyte ratio. One representative experiment of 3 is shown. (B) Similar experiment as panel A, whereby OT-1 splenocytes were activated by OVA in the presence of various amounts of purified EG7 MO-MDSCs or PMN-MDSCs. One representative experiment of 8 is shown. (C) Percentage suppression induced by EG7 MO-MDSC and PMN-MDSC fractions at a 1:1 MDSC/splenocyte ratio, as observed in 8 individual experiments. (D) MO- and PMN-MDSCs were purified from EG7 tumor–bearing β2-microglobulin–deficient C57BL/6 mice and added to OT-1 cultures together with OVA. Percentage suppression represents the mean of 3 individual experiments. (E) OT-1 splenocytes were stimulated with OVA in the presence of BW-Sp3–induced AKR MO- or PMN-MDSCs (1:1 ratio) and percentage suppression was calculated. One representative experiment of 2 is shown. *P < .05; **P < .01.
MO- and PMN-MDSCs suppress antigen-specific T-cell responses via distinct mechanisms, activated through different signaling pathways
Previous studies have suggested that IFN-γ is important for activating the suppressor function of MDSCs.3,8 We therefore investigated whether IFN-γ played a role in the suppression of antigen-specific T-cell responses, mediated by the individual MDSC subfractions. Adding blocking anti–IFN-γ antibodies to OVA-stimulated OT-1 cultures partially reversed T-cell suppression by MO-MDSCs and almost completely reversed suppression by PMN-MDSCs (Figure 5A). This suggests the involvement of IFN-γ–regulated genes in the antiproliferative effect of these tumor-induced myeloid cells.

Both MDSC fractions suppress antigen-driven T-cell responses via an IFN-γ–regulated mechanism, although using distinct signaling pathways and effector molecules. (A) OT-1 splenocytes were stimulated with OVA in the presence of EG7 MO- or PMN-MDSCs (1:1 ratio), with or without 10 μg/mL blocking anti–IFN-γ mAb. One representative experiment of 3 is shown. (B) OT-1 splenocytes were stimulated with OVA in the presence of EG7 MO- or PMN-MDSCs (1:1 ratio) with or without the indicated inhibitors. Values represent the mean plus or minus SD of the relative percentage suppression taken over 4 individual experiments (relative percentage suppression = suppression relative to the suppression exerted by MDSCs without the addition of inhibitors, which is arbitrarily set to 100%; “Methods”). (C) OT-1 splenocytes were stimulated with OVA in the presence of EG7 MO- or PMN-MDSCs (1:1 ratio), and at the end of the culture (42 hours) supernatant was collected and the nitrite concentration was measured. (D,E) MO- and PMN-MDSCs were purified from WT, iNOS−/− (D), or STAT1−/− (E) C57BL/6 mice, bearing EG7 tumors with similar diameters. Percentage suppression of OT-1 proliferation, stimulated with OVA in the presence of various amounts of MDSCs was calculated. Values of percentage suppression represent the mean of 4 (iNOS−/−) and 6 (STAT1−/−) individual experiments. *P < .05; **P < .01; ns indicates not significant.
Both MDSC fractions suppress antigen-driven T-cell responses via an IFN-γ–regulated mechanism, although using distinct signaling pathways and effector molecules. (A) OT-1 splenocytes were stimulated with OVA in the presence of EG7 MO- or PMN-MDSCs (1:1 ratio), with or without 10 μg/mL blocking anti–IFN-γ mAb. One representative experiment of 3 is shown. (B) OT-1 splenocytes were stimulated with OVA in the presence of EG7 MO- or PMN-MDSCs (1:1 ratio) with or without the indicated inhibitors. Values represent the mean plus or minus SD of the relative percentage suppression taken over 4 individual experiments (relative percentage suppression = suppression relative to the suppression exerted by MDSCs without the addition of inhibitors, which is arbitrarily set to 100%; “Methods”). (C) OT-1 splenocytes were stimulated with OVA in the presence of EG7 MO- or PMN-MDSCs (1:1 ratio), and at the end of the culture (42 hours) supernatant was collected and the nitrite concentration was measured. (D,E) MO- and PMN-MDSCs were purified from WT, iNOS−/− (D), or STAT1−/− (E) C57BL/6 mice, bearing EG7 tumors with similar diameters. Percentage suppression of OT-1 proliferation, stimulated with OVA in the presence of various amounts of MDSCs was calculated. Values of percentage suppression represent the mean of 4 (iNOS−/−) and 6 (STAT1−/−) individual experiments. *P < .05; **P < .01; ns indicates not significant.
Well known IFN-γ–regulated genes with a potentially negative impact on T-cell activation include iNOS (NO production), COX-2 (prostaglandin production), and gp91phox (subunit of the ROS-producing NADPH oxidase).32-34 In addition, iNOS, in concert with arginase-1, has been reported as a major contributor to the production of T cell–suppressive RNOSs by MDSCs.20 In an attempt to unravel the suppressive mechanism of the MDSC subfractions, specific inhibitors of each pathway were used in OVA-stimulated cultures (Figure 5B). The iNOS inhibitor L-NMMA was able to partly reverse suppression by MO-MDSCs (on average reducing suppression by 55% ± 5%), suggesting an NO-dependent mechanism, while it did not affect suppression by PMN-MDSCs. Whereas the arginase inhibitor nor-NOHA had no influence on the suppressive activity of MO-MDSCs, it decreased suppression by PMN-MDSCs to some extent in some, but not all, experiments (on average reducing suppression by 24% ± 14%, which did not reach statistical significance). Thus, while arginase activity might have some role in PMN-MDSC–mediated suppression, it is certainly not the main actor in this particular setting. For both subfractions, combining nor-NOHA with L-NMMA did not result in any additional effect. ROSs did not seem to play a role in the suppressive mechanism of either of the MDSC subfractions, as suggested by the inability of the ROS-scavenging enzymes catalase and superoxide dismutase (SOD) to reverse suppression. Inhibiting COX-2–mediated prostaglandin production with the specific COX-2 inhibitor SC-791 did not alter suppression by PMN-MDSCs. However, this inhibitor consistently had a minor effect on MO-MDSC–mediated suppression (on average reducing suppression by 15% ± 6%). Coinhibiting COX-1 function did not further decrease suppression (data not shown), suggesting that prostaglandin production in general is not a major contributor to the MO-MDSC–mediated suppressive activity.
We next corroborated the role of NO in MO-MDSC–mediated suppression. Substantially higher amounts of nitrite could be detected in OVA-stimulated OT-1 cultures containing MO-MDSCs compared with cultures containing no MDSCs or PMN-MDSCs (Figure 5C). To firmly establish the importance of iNOS in the suppressor function of MO-MDSCs, EG7 tumors were grown in iNOS−/− mice, and the respective splenic MDSC fractions were purified and added to OVA-stimulated OT-1 cultures (Figure 5D). In agreement with the effect of L-NMMA on MO-MDSC–mediated suppression, MO-MDSCs originating from iNOS−/− mice showed a reduced suppressive potential compared with WT MDSCs, especially at lower MDSC/splenocyte ratios (1:4, 1:2). However, the ability of L-NMMA to only partially reverse suppression and the substantial residual suppression observed with iNOS−/− MO-MDSCs indicate the existence of additional NO-independent suppressive mechanisms. The suppressor function of PMN-MDSCs, however, does not seem to rely on iNOS or NO production at all.
IFN-γ–mediated gene regulation occurs mainly through STAT1 signaling, although STAT1-independent pathways have been reported.35,36 To evaluate the role of STAT1 in the IFN-γ–driven suppression by MO- and PMN-MDSCs, EG7 tumors were grown in STAT1−/− mice, and the suppressive potential of the respective splenic MDSC subfractions was assessed (Figure 5E). In agreement with the partial dependence on IFN-γ, suppression by MO-MDSCs was significantly reduced in the absence of STAT1. Strikingly, however, the almost entirely IFN-γ–dependent PMN-MDSC–mediated suppression was maintained at the same level by STAT1−/− PMN-MDSCs.
Together, these data reveal functional and mechanistic differences between MDSC subfractions, which would have been obscured by studying total CD11b+Gr-1+ MDSCs.
Immature MO-MDSCs are potential progenitors of strongly suppressive macrophages
CD11b+Gr-1+ cells may possess an internal differentiation/maturation program, which can be switched on in an in vivo environment such as the tumor.37 In addition, such MDSC-derived mature myeloid cells might possess a very potent antiproliferative capacity.12 Neither BW-Sp3 nor EG7 freshly purified total MDSCs were able to suppress anti-CD3–induced proliferation of AKR or C57BL/6 T cells in short-term cultures (Figure 6A). However, when BW-Sp3 MDSCs and naive splenocytes were first coincubated for 2 days, prior to adding anti-CD3 (long-term culture), a profound dose-dependent suppression of T-cell proliferation was observed, which was already prominent at a 1:16 MDSC/splenocyte ratio (Figure 6A). Interestingly, only purified BW-Sp3 MO-MDSCs, but not PMN-MDSCs, can recapitulate this suppression, indicating that the suppressor cells are strictly MO-MDSC derived (Figure 6B). Remarkably, such suppression was never observed in long-term cultures containing EG7 total MDSCs (Figure 6A) or their individually purified subpopulations (Figure 6B).

The MO-MDSC fraction of BW-Sp3, but not EG7, tumor–bearing mice gives rise to macrophages able to suppress polyclonal T-cell activation via NO. (A) Total CD11b+Gr-1+ MDSCs were purified from BW-Sp3 or EG7 tumor-bearers and added in various amounts to naive AKR or C57BL/6 splenocytes, respectively. These cocultures were stimulated with 1 μg/mL anti-CD3, either following the “short-term culture” or “long-term culture” protocol (“Methods”). (B) BW-Sp3 and EG7 MO- and PMN-MDSC fractions were purified and added in the indicated amounts to naive AKR or C57BL/6 splenocytes, respectively. The experiment was executed according to the long-term culture protocol. (C) Supernatant was collected at the end of the culture and nitrite concentrations were determined. (D) Purified BW-Sp3 MO-MDSCs were added at a 1:1 ratio to naive AKR splenocytes and stimulated following the long-term culture protocol in the presence or absence of the indicated inhibitors. (E) At the end of the long-term culture, plastic nonadherent cells were thoroughly washed away and pictures from the adherent cells were taken. Adherent cells were collected by gentle scraping, and expression of the indicated markers was analyzed by flow cytometry. The percentage of CD11b+F4/80+ mature macrophages (gate R1) in each adherent cell population is indicated. Histograms represent gated CD11b+ cells stained with isotype control mAbs (dotted line) or marker-specific mAbs (full line). (F) Purified BW-Sp3 MO-MDSCs were added at different ratios to naive AKR splenocytes and cultured for 3 days. Subsequently, anti-CD3 was added to either undisturbed cultures (= total: adherent + nonadherent cells) or to the plastic nonadherent fraction of the cultures, which was transferred into fresh wells (= nonadherent cells). Twenty-four hours later, 3H-thymidine was provided and was allowed to incorporate for another 18 hours. *P < .05; **P < .01; NS indicates not significant. One representative experiment of at least 3 is shown.
The MO-MDSC fraction of BW-Sp3, but not EG7, tumor–bearing mice gives rise to macrophages able to suppress polyclonal T-cell activation via NO. (A) Total CD11b+Gr-1+ MDSCs were purified from BW-Sp3 or EG7 tumor-bearers and added in various amounts to naive AKR or C57BL/6 splenocytes, respectively. These cocultures were stimulated with 1 μg/mL anti-CD3, either following the “short-term culture” or “long-term culture” protocol (“Methods”). (B) BW-Sp3 and EG7 MO- and PMN-MDSC fractions were purified and added in the indicated amounts to naive AKR or C57BL/6 splenocytes, respectively. The experiment was executed according to the long-term culture protocol. (C) Supernatant was collected at the end of the culture and nitrite concentrations were determined. (D) Purified BW-Sp3 MO-MDSCs were added at a 1:1 ratio to naive AKR splenocytes and stimulated following the long-term culture protocol in the presence or absence of the indicated inhibitors. (E) At the end of the long-term culture, plastic nonadherent cells were thoroughly washed away and pictures from the adherent cells were taken. Adherent cells were collected by gentle scraping, and expression of the indicated markers was analyzed by flow cytometry. The percentage of CD11b+F4/80+ mature macrophages (gate R1) in each adherent cell population is indicated. Histograms represent gated CD11b+ cells stained with isotype control mAbs (dotted line) or marker-specific mAbs (full line). (F) Purified BW-Sp3 MO-MDSCs were added at different ratios to naive AKR splenocytes and cultured for 3 days. Subsequently, anti-CD3 was added to either undisturbed cultures (= total: adherent + nonadherent cells) or to the plastic nonadherent fraction of the cultures, which was transferred into fresh wells (= nonadherent cells). Twenty-four hours later, 3H-thymidine was provided and was allowed to incorporate for another 18 hours. *P < .05; **P < .01; NS indicates not significant. One representative experiment of at least 3 is shown.
BW-Sp3 MO-MDSC–induced suppression coincided with the appearance of significant nitrite concentrations in the cultures, indicative of high NO production (Figure 6C). Nitrite accumulation was not seen in the presence of nonsuppressive EG7 MDSCs (Figure 6C). The addition of L-NMMA almost completely restored T-cell proliferation in cultures containing BW-Sp3 MDSCs (Figure 6D), indicating that suppression was NO mediated. Inhibiting arginase activity did not significantly alter MDSC-mediated suppression. In addition, blocking anti–IFN-γ antibodies partially reversed T-cell suppression, in accordance with the role of this cytokine in regulating iNOS gene expression.
We next wondered whether the strong BW-Sp3 MO-MDSC–dependent antiproliferative effect upon long-term culture could be mediated by a more differentiated cell type. In this context, both in BW-Sp3 and EG7 total MDSC- or MO-MDSC–supplemented cultures a gradual accumulation of plastic-adherent cells was observed from day 2 onward (Figure 6E), while such cells were not seen in cultures containing PMN-MDSCs or cultures without MDSCs added (data not shown). FACS analysis showed that the BW-Sp3 MO-MDSC–derived cells consisted predominantly (> 90%) of mature CD11bhighF4/80highMHC II+ macrophages, which only marginally expressed Gr-1 and no Ly6G (Figure 6E). Importantly, T-cell proliferation was not suppressed if the plastic-nonadherent cells were separated from the adherent fraction and subsequently activated with anti-CD3 (Figure 6F). This indicates that suppression was solely mediated by these adherent macrophages. Remarkably, macrophage homogeneity was lower for EG7 plastic-adherent cells (50%-60%; Figure 6E), suggesting that EG7 MO-MDSCs had a lower capacity to differentiate toward mature macrophages under the given culture conditions.
These results show that MO-MDSCs might possess suppressive activity at 2 levels, either as immature cells or as differentiated macrophages.
The differences in MDSC differentiation observed between EG7 and BW-Sp3 are tumor driven
Finally, we wondered whether the differences between EG7 and BW-Sp3 MO-MDSCs were a tumor-driven phenomenon, or whether these were intrinsic differences between C57BL/6 and AKR monocytes, irrespective of the tumors. To discriminate between these possibilities, EG7 and BW-Sp3 tumors were grown in (AKR × C57BL/6)F1 mice. Both EG7 and BW-Sp3 F1 PMN- and MO-MDSCs had a similar surface marker profile as their C57BL/6 or AKR counterparts (data not shown), and—in line with previous results—dose-dependently suppressed OT-1 T-cell responses in a short-term assay (Figure 7A), the MO-MDSC fraction through a partly NO-mediated mechanism (Figure 7B). Next, F1 MDSCs were assessed for their suppressive activity toward anti-CD3–stimulated F1 splenocytes in a long-term assay. Interestingly, while EG7 F1 MDSCs mice did not influence polyclonal T-cell activation, F1 BW-Sp3 MDSCs were again suppressive (Figure 7C). As anticipated, only BW-Sp3–induced F1 MO-MDSCs were able to recapitulate this suppression (Figure 7D). These results indicate that, in a given genetic background, distinct tumors can differentially regulate MDSC differentiation.

Differences in the ability of MDSCs to suppress polyclonal T-cell activation are tumor driven. (A) OT-1 splenocytes were stimulated with OVA in the presence of BW-Sp3– or EG7-induced F1 MO- or PMN-MDSCs (1:1 ratio). One representative experiment of 2 is shown. (B) OT-1 splenocytes were stimulated with OVA in the presence of EG7 F1 MO- or PMN-MDSCs (1:1 ratio) with or without 0.5 mM L-NMMA. One representative experiment of 2 is shown. *P < .05 (C) BW-Sp3 and EG7 tumors were grown in (AKR × C57BL/6)F1 mice and total MDSCs were purified from mice with similar tumor diameters. Subsequently, BW-Sp3– and EG7-induced F1 MDSCs were added in various amounts to naive F1 splenocytes, which were subjected to the long-term culture protocol. One representative experiment of 3 is shown. (D) MO- and PMN-MDSCs were individually purified from BW-Sp3 tumor–bearing F1 mice and were added in various amounts to naive F1 splenocytes, which were subjected to the long-term culture protocol.
Differences in the ability of MDSCs to suppress polyclonal T-cell activation are tumor driven. (A) OT-1 splenocytes were stimulated with OVA in the presence of BW-Sp3– or EG7-induced F1 MO- or PMN-MDSCs (1:1 ratio). One representative experiment of 2 is shown. (B) OT-1 splenocytes were stimulated with OVA in the presence of EG7 F1 MO- or PMN-MDSCs (1:1 ratio) with or without 0.5 mM L-NMMA. One representative experiment of 2 is shown. *P < .05 (C) BW-Sp3 and EG7 tumors were grown in (AKR × C57BL/6)F1 mice and total MDSCs were purified from mice with similar tumor diameters. Subsequently, BW-Sp3– and EG7-induced F1 MDSCs were added in various amounts to naive F1 splenocytes, which were subjected to the long-term culture protocol. One representative experiment of 3 is shown. (D) MO- and PMN-MDSCs were individually purified from BW-Sp3 tumor–bearing F1 mice and were added in various amounts to naive F1 splenocytes, which were subjected to the long-term culture protocol.
Discussion
In recent years, multiple reports have shown that myeloid cells with immune-suppressive activity, also known as MDSCs, can accumulate during cancer progression and inhibit antitumor T-cell responses.2-10 However, the term MDSC covers a heterogeneous population of cells, which in mice are characterized by CD11b and Gr-1 coexpression. For a thorough understanding of the biology and activities of these cells, it is crucial to tackle their heterogeneity and to evaluate the origin and function of different MDSC subpopulations. In this study, our data firmly establish the coexistence of functionally distinct mononuclear and polymorphonuclear lineages within tumor-induced splenic MDSCs, and largely extend their surface marker profile, including markers with MO-MDSC/PMN-MDSC discriminating potential.
MO-MDSCs are typified by the monocyte/macrophage lineage markers F4/80 and CD115, but also other MO-MDSC markers are in conformity with the reported phenotype of peripheral blood monocytes (CD31+LFA1+VLA-4+CD16/32+CD44+MHC II−).25 In particular, MO-MDSCs share the major characteristics of the “inflammatory” subset of monocytes (Ly6ChighCCR2+CX3CR1−/lowCD62L+), which have a high potential to migrate to inflammatory sites.25,26 PMN-MDSCs had a high SSC profile and expressed the typical neutrophil marker Ly6G.28 For mouse peripheral blood neutrophils, biochemically and functionally distinct subsets have been characterized in a systemic inflammatory setting as being CD11b−VLA-4+ (PMN-I) or CD11b+VLA-4− (PMN-II).38 The splenic PMN-MDSCs from tumor-bearing mice did not fit into this classification, considering their combined expression of CD11b and VLA-4 (Table 2). Furthermore, PMN-MDSCs were low-density cells. Up to now, such a low-density phenotype has been reported mainly for human granulocytes upon G-CSF treatment or fMLP activation and in patients confronted with overwhelming inflammation or cancer.39-41
While in naive mice CD11b+Ly6C+Ly6G− splenocytes consisted mainly of monocytes and eosinophils; the latter did not expand in tumor-bearers. Furthermore, the phenotype of tumor-induced monocytes was not identical to that of their naive counterparts, expressing higher levels of CD71, CD115, and CD80. Remarkably, CD80 was recently shown to be important for the function of MDSCs.42
Both MO- and PMN-MDSCs were shown to efficiently suppress antigen-driven CD8+ T-cell proliferation in short-term culture assays. While T-cell suppression by granulocytic cells has been reported in human cancer patients,7,39 this is the first study showing that PMN cells, with a potential T cell–suppressive activity, are expanded in tumor-bearing mice. In this context, a recent report indicated that G-CSF treatment can induce low-density granulocytes, which inhibit acute graft-versus-host disease in mice.43
We wondered whether the suppression mediated by tumor-induced MDSCs relied on the induction of T-cell anergy through an MHC-restricted presentation of antigen, as described by others.4,6 By assessing the suppressive activity of MHC I–deficient or allogeneic MDSCs, this was shown to be unlikely in our in vitro cultures. However, we do not exclude that antigen-specific induction of T-cell anergy might be an additional mechanism used by MDSCs in vivo. In addition, it is plausible that MDSCs might indirectly suppress antigen-specific T-cell activation by inhibiting the function of antigen-presenting cells (APCs). In this regard, a recent study has suggested the existence of an MDSC-macrophage cross talk resulting in increased IL-10 secretion, redirecting macrophage activation.44 We investigated whether a similar cross talk could be at work between MDSCs and splenic dendritic cells (DCs), which are the APCs most likely responsible for activating T cells in OT-1 cultures. In favor of a DC:MDSC cross talk, our preliminary data show that coculturing purified splenic DCs with MDSCs in the presence of LPS/IFN-γ can induce significantly higher amounts of IL-10 compared with the sum of IL-10 produced by DCs or MDSCs alone (K.M., unpublished observation, September 2007).
Several studies have indicated that IFN-γ is important for activating MDSC-mediated suppression.3,8 In accordance with this, we found that IFN-γ blocking partially reduced suppression by MO-MDSCs and almost completely abolished suppression by PMN-MDSCs. IFN-γ can affect gene transcription through STAT1-dependent or -independent signaling pathways.35,36 Interestingly, in this context, our data uncovered a fundamental difference between MO-MDSCs, which are partially dependent on STAT1 for their suppressive activity, and PMN-MDSCs, which are not. Furthermore, MO-MDSCs use a STAT1-dependent gene such as iNOS45 for suppression, as evidenced by a partial impairment of the suppressive activity in iNOS−/− MO-MDSCs or upon administration of the iNOS inhibitor L-NMMA. iNOS-generated effector molecules are either NO or ROS/RNOS, the latter of which is produced in conditions of high arginase-1 activity.20 In the present study, NO most likely mediates suppression, as indicated by (1) the significantly enhanced NO2− concentrations in the supernatant, (2) the inability of an arginase inhibitor to further enhance the effect of L-NMMA, and (3) the inability of superoxide dismutase or catalase to diminish suppression. A significant portion of MO-MDSC–mediated suppression is IFN-γ/STAT1/iNOS independent, warranting further research to uncover the molecules involved.
As a second point of divergence between MDSC subfractions, iNOS is not implicated in PMN-MDSC–mediated suppression. Also ROS and arginase, though reported as potential PMN-suppressor molecules,7,39 play no major role. The unimpaired suppression by STAT1−/− PMN-MDSCs is remarkable, considering their entirely IFN-γ–dependent suppressive mechanism, and suggests that the suppressive mediator might be one of the described IFN-γ–induced/STAT1-independent factors.35,36 Alternatively, it has been shown that in STAT1-deficient cells, IFN-γ can induce a set of genes that are not transcribed in WT cells and are hence not truly STAT1 independent.35 Therefore, we cannot exclude that in STAT1−/− PMN-MDSCs other suppressive mechanisms, normally not seen in WT mice, are at work. Nevertheless, in favor of a STAT1-independent mechanism are data describing that immature or GM-CSF–treated human neutrophils—2 populations that possibly resemble PMN-MDSCs in mice—contain lower levels of STAT1 that cannot be tyrosine-phosphorylated upon IFN-γ stimulation.46
While MDSCs can affect T-cell responses as immature cells in the peripheral lymphoid organs, they can also differentiate into more mature cell types with tumor-promoting effects. Indeed, MDSCs can be progenitors of tumor-associated macrophages (TAMs) in vivo37 and alternatively activated macrophages in vitro.12 Moreover, MDSCs are able to promote tumor angiogenesis by differentiating into endothelial cells in the tumor microenvironment.47 However, it remains to be clarified whether tumors regulate the MDSC differentiation program and which subpopulations are involved. We showed that upon prolonged in vitro MDSC/splenocyte coculture, BW-Sp3–induced MO-MDSCs differentiated into highly suppressive mature macrophages, which inhibited anti-CD3–induced (or ConA-induced; data not shown) T-cell proliferation in an NO-dependent fashion. Remark that this differentiation did not require the supplementation of growth factors to the culture. An important consequence of these data are the notion that for the study of MDSCs in vitro, any protocol taking longer than 48 hours might reflect MDSC-derived macrophage functions, rather than functions of the immature/undifferentiated MDSCs found in the peripheral organs. Strikingly, macrophage differentiation was less efficient for EG7 MO-MDSCs, which also did not suppress polyclonal T-cell responses. Interestingly, expression of the immature myeloid marker Sca-1,24 on MO-MDSCs from BW-Sp3 but not EG7 tumor-bearers, suggests a difference in the maturation state of these MDSC fractions and provides a possible explanation for their distinct in vitro differentiation potential. Furthermore, Sca-1 expression was a tumor-induced phenomenon and was not found on CD11b+Gr-1+ cells from naive AKR mice.
To obtain MDSCs from an identical genetic background, we injected BW-Sp3 and EG7 in (AKR × C57BL/6)F1 mice. In this given background, we found that the functional differences in EG7 and BW-Sp3 MDSCs were tumor driven. However, these data do not exclude the possibility that EG7 and BW-Sp3 instruct different characteristics onto MDSCs in mice with a somewhat altered genetic program and a different combination of alleles. Nonetheless, these data allow to conclude that, besides a potential impact of genetic background, there is definitely an important influence of the tumor on MDSC characteristics. Various tumor-derived factors (TDFs) have been described to contribute to MDSC recruitment and function, several of which provoke an MDSC differentiation blockade.13-18,48,49 Possibly, variations in such TDFs could be responsible for the differences in maturation state and differentiation potential between EG7 and BW-Sp3 MDSCs. In the same vein, it was shown that IL-1β–transfected 4T1 mammary carcinoma tumors induce MDSCs that are phenotypically and functionally distinct from MDSCs elicited by nontransfected tumors.16
Overall, current data establish distinctive mononuclear and polymorphonuclear tumor-induced myeloid cells as contributors to immune unresponsiveness in cancer and provide novel insights in their molecular and functional complexity.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Ella Omasta, Marie-Thérèse Detobel, and Eddy Vercauteren for excellent technical assistance.
This work was supported by a doctoral grant from the Fund for Scientific Research Flanders (FWO-Vlaanderen) (K.M., M.G., and J.V.B.); a doctoral grant from the Institute for Promotion and Innovation by Science and Technology in Flanders (IWT-Vlaanderen; R.V.B.); and a postdoctoral grant from the Stichting tegen Kanker (J.A.V.G.).
Authorship
Contribution: K.M. designed and performed research, analyzed and interpreted data, and drafted the paper; M.G., J.V.B., and R.V.B. performed research; C.G. provided materials; A.B. and P.D.B. designed research; J.A.V.G. designed and performed research, interpreted data, and drafted the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jo A. Van Ginderachter, Lab of Cellular and Molecular Immunology, Vrije Universiteit Brussel, Building E, Level 8, Pleinlaan 2, B-1050 Brussels, Belgium; e-mail: jvangind@vub.ac.be.
