

Introduction
In its August 15, 2007, issue, Blood published a proposal for revision of the World Health Organization (WHO) diagnostic criteria for the chronic myeloproliferative disorders (MPDs) polycythemia vera (PV), essential thrombocytosis (ET), and primary myelofibrosis (PMF).1 Algorithms based on these diagnostic criteria were subsequently published in Leukemia.2 Ostensibly prompted by newly described MPD molecular abnormalities, the proposed revision was both timely and appropriate. The initial WHO diagnostic criteria for these disorders, published in 2001,3 were never prospectively evaluated and subsequently were invalidated.4 The discovery of JAK25-9 and MPL10-12 gene mutations not only provided new insights into the molecular basis of the MPD but also new molecular approaches to their diagnosis. Unfortunately, the proposed guideline revision and the attendant algorithms not only recapitulated all the faults of the initial WHO diagnostic criteria but also failed to capitalize on the biologic insights and opportunities offered by these newly discovered mutations to improve diagnostic accuracy. This was because the proposed revision eschewed the fundamental tenets of evidence-based medicine.13-15 The purpose of this review, therefore, is to offer an alternative perspective of the diagnostic approach to PV, ET, and PMF to enable clinicians to select the appropriate diagnostic tests for a particular MPD within the context of their own practices.
The back story
The MPDs are neither new nor rare diseases, but they continue to confound physicians diagnostically for reasons that are many and cogent. Although not rare, the MPDs are sufficiently uncommon that most physicians see few such patients; and because disease duration for the MPD is typically measured in decades, physicians rarely have the opportunity to observe their full natural history. Importantly in this regard, the initial clinical manifestations of the MPDs are highly variable and their clinical phenotypes are also subject to change with time. These disorders not only mimic each other phenotypically but many other benign and malignant blood disorders as well. For example, PV can present as isolated erythrocytosis,16 leukocytosis,17 thrombocytosis18,19 (Figure 1A), or even myelofibrosis,20,21 whereas isolated thrombocytosis is the presenting feature in approximately 20% of PMF patients.22 In addition, myelofibrosis is a well-recognized feature of PV23-25 and erythrocytosis can develop in PMF during the course of the illness26 (Figure 1B).

Evolution of essential thrombocytosis and primary myelofibrosis into polycythemia vera. (A) Erythrocytosis developing in a 60-year-old man with essential thrombocytosis 6 years after diagnosis. The increase in the JAK2 V617F neutrophil allelic burden with time is also shown. The hemoglobin (Hgb) level was reduced by phlebotomy. (B) Erythrocytosis developing in a 70-year-old woman with classic PMF of 17 years' duration, while taking hydroxyurea to control splenic enlargement (bracketed line). The hemoglobin (Hgb) level was reduced by phlebotomy.
Evolution of essential thrombocytosis and primary myelofibrosis into polycythemia vera. (A) Erythrocytosis developing in a 60-year-old man with essential thrombocytosis 6 years after diagnosis. The increase in the JAK2 V617F neutrophil allelic burden with time is also shown. The hemoglobin (Hgb) level was reduced by phlebotomy. (B) Erythrocytosis developing in a 70-year-old woman with classic PMF of 17 years' duration, while taking hydroxyurea to control splenic enlargement (bracketed line). The hemoglobin (Hgb) level was reduced by phlebotomy.
William Osler was not the first to identify PV as a distinct clinical entity,27 but he was the first to recognize its capacity for phenotypic mimicry and devised diagnostic criteria that addressed the problem.28 The Polycythemia Vera Study Group (PVSG) subsequently expanded Osler's diagnostic criteria29 and, as new knowledge was obtained, other groups formulated diagnostic criteria for PV,30,31 ET,32 and PMF,33 but to date there has been no uniformly agreed on set of diagnostic criteria, or at least not one, according to recent surveys,34,35 to which most clinicians strictly adhere.
In 2001, the WHO attempted to fill this void with a new MPD classification, grouping PV, ET, and PMF together with chronic myelogenous leukemia, chronic neutrophilic leukemia, chronic eosinophilic leukemia, the hypereosinophilic syndrome, and unclassifiable MPDs under the rubric of “the chronic myeloproliferative diseases.”3,36 The rationale was to apply to the MPD the WHO Revised European-American Lymphoma (REAL) classification paradigm that had been successfully used for lymphoid and myeloid neoplasms. The REAL scheme combines morphology, genotype, immunophenotype, and clinical phenotype to define distinct clinical entities.3 The principles espoused by the WHO were both laudable and appropriate, but only to the extent that the REAL classification paradigm used for other hematologic neoplasms was applicable to the MPD; unfortunately, this proved to be limited. The WHO also provided diagnostic criteria for the MPDs, and these soon proved to be problematic.4
The problem
The WHO MPD diagnostic classification was based on the shared features of “myeloproliferation” with relatively normal maturation and a tendency to extramedullary hematopoiesis that characterize the various MPDs.3 The shared feature of myeloproliferation, however, was more apparent than real because, with respect to the involved stem cell, PV, ET, and PMF are actually disorders of myeloaccumulation, not myeloproliferation.37-39 In this regard, in contrast to the other “myeloproliferative” disorders, survival with PV, ET, or PMF, even with supportive therapy alone, is usually measured in decades, and transformation to acute leukemia is much less common and often treatment-related. Beyond these characteristics, PV, ET, and PMF share more in common genotypically and phenotypically with each other than they do with the other MPDs with which they have been classified36 ; and on this basis alone, they merited a separate classification. This contention was solidified recently by the discovery of JAK25-9 and MPL gene mutations10-12 in MPD patients. Indeed, considering their genotypic similarities and the tendency for each disorder to acquire the phenotypic characteristics of the others, it is worth asking whether PV, ET, and PMF are separate diseases, different manifestations of the same disease, or a combination of both, and current molecular evidence supports the last possibility.22,40,41
The REAL paradigm, although useful for distinguishing and classifying lymphoid and many myeloid neoplasms, was not appropriate for PV, ET, and PMF. These 3 disorders share in common the following features: origin in a multipotent hematopoietic progenitor cell, relatively normal cellular maturation, phenotypic and genotypic mimicry, and a tendency to evolve into each other or develop myelofibrosis. They also do not have unique immunophenotypes. Therefore, none of the REAL paradigm tenets, alone or together, was diagnostically useful.
Nevertheless, the WHO based their diagnostic criteria on morphology, and with respect to PV, as a surrogate for red cell mass and plasma volume studies, substituted hemoglobin values of more than 18.5 g/dL in men and more than 16.5 g/dL in women, or more than 99th percentile of the chosen method-specific reference range for age, sex, and altitude of residence.3 However, no data were offered to support the substitution of specific hemoglobin values as a surrogate for direct measurement of the red cell mass. Ostensibly, the hemoglobin reference standards were for those physicians who lacked access to a nuclear medicine facility, but proof that such values were clinically meaningful was not provided.
Considering the many phenotypic similarities between PV, ET, and PMF, what should have been most important was defining their differences. With respect to laboratory characteristics, only erythrocytosis sets PV apart from its companion MPD, whereas during the early stages of the disease, an increase in circulating CD34+ cells is characteristic for PMF, although this is not uniformly so.42,43 Importantly, ET has no specific clinical or laboratory characteristics that distinguish it from PV. Therefore, considering that trilineage involvement is the ultimate possible phenotype for an MPD arising in a multipotent hematopoietic stem cell, recommendation of an accurate method for detecting absolute erythrocytosis should have been mandatory. Unfortunately, the WHO alternatives to direct measurement of the red cell mass proved to be inadequate because of the erroneous assumption that, in an MPD patient, a normal hemoglobin or hematocrit level signified that the red cell mass was normal.
In 2005, Johansson et al4 challenged the WHO assertion that only hemoglobin levels greater than 18.5 g/dL (hematocrit > 55.5%) in a man or greater than 16.5 g/dL (hematocrit > 49.5%) in a woman (or the equivalent > 99th percentile of the method-specific reference value) established the presence of absolute erythrocytosis. They applied the WHO criteria to 77 PV patients and 66 patients with apparent erythrocytosis, all of whom had direct red cell mass and plasma volume measurements.4 They found that the WHO criteria identified absolute erythrocytosis in only 35% of the male PV patients and 63% of the women, whereas 14% of the men and 35% of the women without erythrocytosis were noted as having it (Figure 2).

Correlation of the WHO hemoglobin guidelines for the diagnosis of PV with actual red cell mass and plasma volume measurements. (A) Men. (B) Women. Similar results were obtained if the corresponding hematocrit values were used. The data are recalculated from Johansson et al.4
Correlation of the WHO hemoglobin guidelines for the diagnosis of PV with actual red cell mass and plasma volume measurements. (A) Men. (B) Women. Similar results were obtained if the corresponding hematocrit values were used. The data are recalculated from Johansson et al.4
Moreover, the degree to which the WHO hemoglobin criteria failed was actually worse than it appeared because, by direct measurement, the red cell mass is not considered elevated unless it is 125% of normal.44 This not only enhances the specificity of the test, it also indicated that the WHO recommendations lacked sensitivity. Surprisingly, these objective data were omitted in the revised WHO diagnostic recommendations, which remain unchanged.1 The specificity of the WHO PV diagnostic criteria was also challenged by the British Committee for Standards in Haematology.45
Blood volume physiology
Most discussions of the blood volume emphasize the important direct and exponential relationship between hematocrit and blood viscosity as it occurs in large vessels.46,47 However, the emphasis should really be on the behavior of blood flow in arterioles, capillaries, and venules. In these small vessels, the ratio of vessel surface area to its volume is greatest and the exposure of the blood to the frictional drag of the vessel wall is maximal. Thus, the flow of plasma nearest the vessel wall is retarded compared with the flow of red cells at the vessel center.48 Because of this, there are always fewer red cells in these small vessels; and as a consequence, the volume of distribution of red cells in the circulation differs from that of plasma. Because the microvasculature composes almost 20% of the circulatory system,49 the hematocrit of blood taken from a peripheral artery or vein will not accurately reflect the total body hematocrit.50
From a practical perspective, this has important ramifications with respect to the potential for organ-specific thrombosis when the red cell mass is increased. First, the hematocrit is not uniform in all organs, being highest in the spleen and liver and lowest in the brain, bowel, and kidneys.49 Second, normally, whenever there is a hypoxia-induced increase in erythropoiesis, there is a reciprocal decrease in the plasma volume.51-54 This is also true when red cell transfusions are given55 and has the effect of maintaining a normal blood volume at the expense of an increase in peripheral vascular resistance (Table 1). In PV, however, the plasma volume usually does not shrink with the development of erythrocytosis and may even expand, particularly in women (Table 1), masking the absolute increase in red cell mass.39,56-58 Thus, it is not surprising that the WHO hemoglobin or hematocrit guidelines were invalid.
Examples of the diagnostic value of red cell mass (RCM) and plasma volume (PV) determinations
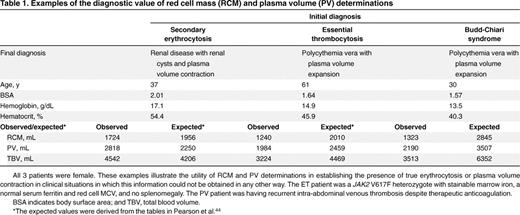
All 3 patients were female. These examples illustrate the utility of RCM and PV determinations in establishing the presence of true erythrocytosis or plasma volume contraction in clinical situations in which this information could not be obtained in any other way. The ET patient was a JAK2 V617F heterozygote with stainable marrow iron, a normal serum ferritin and red cell MCV, and no splenomegaly. The PV patient was having recurrent intra-abdominal venous thrombosis despite therapeutic anticoagulation.
BSA indicates body surface area; and TBV, total blood volume.
*The expected values were derived from the tables in Pearson et al.44
There are also other important implications of red cell mass and plasma volume determinations not addressed by the latest WHO recommendations. First, a high hematocrit is not synonymous with erythrocytosis any more than a normal hematocrit is synonymous with the absence of erythrocytosis when PV is a diagnostic consideration.39 A high hematocrit can be simply the result of plasma volume contraction (Table 1). Indeed, unless the hematocrit is more than or equal to 60% in a man or woman, it is not possible to distinguish plasma volume contraction from absolute erythrocytosis.59 It was for this reason that the PVSG stipulated that direct determination of the red cell mass and plasma volume should be an integral part of the evaluation of a high hematocrit.29 The clinical problem of plasma volume contraction is not trivial,60-63 and there is no excuse for ignoring this group of patients.
We recognize the WHO concern that some physicians may not have access to red cell mass and plasma volume measurements because of economic or geographic considerations. In this regard, the assumption that JAK2 mutation assays are not currently subject to the same constraints is also erroneous. From our perspective, in developed countries, it is a deviation from standard of care if these tests are not available, at the very least, in major academic centers. For other circumstances, we offer some alternatives. First, microcytic erythrocytosis is an important clue to the presence of an increased red cell mass (Figure 3).64 Second, because it is necessary for the red cell mass to be greater than 125% of normal to qualify for absolute erythrocytosis, phlebotomy can be diagnostic as well as therapeutic. If absolute erythrocytosis is suspected, there should be a minimum excess of approximately 700 mL of red cells in either a man or woman. If reduction of the hematocrit to less than 45% in a man or less than 42% in a woman requires 2 or more phlebotomies, absolute erythrocytosis can be assumed.

Comparison of simultaneous hematocrits and red cell counts in a woman with PV and hepatic vein thrombosis experiencing recurrent thromboses of stents and shunts during therapeutic anticoagulation. The red cell count is above normal at the time of each thrombosis, whereas the hematocrit is not. The heavy lines indicate the upper and lower limits of normal for each.
Comparison of simultaneous hematocrits and red cell counts in a woman with PV and hepatic vein thrombosis experiencing recurrent thromboses of stents and shunts during therapeutic anticoagulation. The red cell count is above normal at the time of each thrombosis, whereas the hematocrit is not. The heavy lines indicate the upper and lower limits of normal for each.
A further important PVSG stipulation was that, after such a phlebotomy trial, the hematocrit should increase by at least 10% within 3 months in the absence of iron deficiency.65 These criteria should ensure that physicians without access to a nuclear medicine facility can achieve diagnostic accuracy; they should not, however, be used as surrogates for direct red cell mass and plasma volume measurements when available because not all patients with a high hemoglobin or hematocrit have an elevated red cell mass, whereas many MPD patients with a supposedly normal hematocrit or hemoglobin level do.56,57 Finally, it should be emphasized that red cell mass and plasma volume determinations only establish the presence of erythrocytosis, not its cause.
JAK2 V617F
The discovery of the JAK2 V617F mutation5-9 was the most important advance in the study of the MPD since the demonstration 30 years earlier that these were clonal disorders involving a multipotent hematopoietic stem cell.66,67 JAK2 is the cognate tyrosine kinase of the erythropoietin and thrombopoietin receptors and also the obligate chaperone responsible for their cell surface expression.68,69 The substitution of phenylalanine for valine (V617) in the regulatory JH2 domain of JAK2 leading to constitutive kinase activation occurs in approximately 95% of PV patients and in approximately 50% of PMF and ET patients.41,70,71 Indeed, this mutation explains many of the clinical and laboratory features shared by these 3 disorders, although it does not appear to be the initiating mutation.40,72-74
How one mutation could be responsible for 3 different clinical phenotypes is still unresolved, but in vitro clonal assays,38 animal models,75 and studies quantifying the JAK2 V617F neutrophil allele burden in MPD patients41,76,77 indicate that both gene dosage and sex of the patient have roles. In ET, in which females predominate, the JAK2 V617F neutrophil allele burden is usually low41 and isolated thrombocytosis is the rule, whereas in PV, the higher neutrophil allele burden was associated with higher hematocrit and leukocyte counts, a lower platelet count, splenomegaly, and pruritus.78 As a corollary, some ET patients with a rising neutrophil allele burden transform over time to PV (Figure 1A) or PMF, although JAK2 V617F expression is not mandatory for this to occur.77 Importantly, ET patients expressing JAK2 V617F also appear to have a “PV-like” phenotype compared with their JAK2 V617F− counterparts, even to the extent of having an increased incidence of venous thrombosis.79 Figure 4 illustrates quantitative neutrophil JAK2 V617F allelic burdens in ET, PV, and PMF patients,41 demonstrating not only their important differences but also the significant overlap that occurs, presumably accounting in part for their phenotypic mimicry.

Comparison of the quantitative JAK2 V617F neutrophil allelic burdens in ET, PV, and PMF at diagnosis, illustrating the differences in allelic burden among them. The degree of overlap between the neutrophil allele burden in ET and the other MPD is indicated by the horizontal bars.
Comparison of the quantitative JAK2 V617F neutrophil allelic burdens in ET, PV, and PMF at diagnosis, illustrating the differences in allelic burden among them. The degree of overlap between the neutrophil allele burden in ET and the other MPD is indicated by the horizontal bars.
Given these data, because PV, PMF, and ET all arise in a multipotent hematopoietic stem cell and the JAK2 V617F mutation appears to cause a “PV-like” phenotype in ET patients, it is illogical not to consider the possibility of PV when evaluating a patient with thrombocytosis. Indeed, based on a recent publication by Cassinat et al,80 the new WHO dictum that red cell mass and plasma volume studies are not required in the evaluation of isolated thrombocytosis1 is invalid. These authors found that red cell mass and plasma volume determinations identified erythrocytosis in 46% of patients initially considered to have ET by the WHO hemoglobin criteria, but when the JAK2 V617F mutation was present, the proportion rose to 64%.80 An example of such a patient is provided in Table 1.
Although the WHO correctly concluded that a JAK2 V617F assay is an important diagnostic test in the evaluation of an MPD, no guidance was provided with respect to test sensitivity and specificity. For example, in the Cassinat et al study,80 the JAK2 V617F assay had a sensitivity of 86% when the red cell mass was elevated, but its specificity was only 64%. It is now clear that a positive JAK2 V617F assay is not diagnostic for a particular MPD, and assay-positive patients with isolated erythrocytosis have been identified, who have not evolved to classic PV.12,81 Finally, because JAK2 V617F is responsible for many of the biochemical abnormalities associated with the MPD, it is unclear why the WHO still requires a serum erythropoietin level or the iconic but usually unobtainable endogenous erythroid colony assay for diagnostic purposes if JAK2 V617F is present.2
Bone marrow morphology as a diagnostic test
It is a stated WHO concern that bone marrow morphology was not optimally used as a diagnostic tool by the PVSG.3 However, this concern ignores a careful PVSG analysis of bone marrow morphology in 281 PV patients followed for more than 9 years.20 In this study, 13% of patients did not have increased marrow cellularity at diagnosis; megakaryocyte hyperplasia correlated with marrow cellularity, and 11% of patients had a moderate to marked increase in reticulin early in their disease that had no bearing on prognosis. More than 90% of a parallel series of untreated ET patients had marrow cellularity greater than 50% together with megakaryocyte hyperplasia.82 Thus, the PVSG concluded that marrow morphology was an inadequate tool for distinguishing PV from ET or PMF and stipulated the use of a red cell mass determination for this purpose.29
In 2001, the WHO also endorsed the concept of prefibrotic PMF and provided diagnostic criteria for it,3,36 but no prospective study validating these criteria was ever conducted. Indeed, the proposed morphologic criteria were based entirely on retrospective studies claiming that marrow histology could distinguish the “prefibrotic cellular phase” of PMF from “true” ET.83 Interestingly, when these criteria were applied in an unblinded fashion to 116 ET patients, 70% were reclassified as having PMF, an epidemiologically implausible outcome84 that led to concern about the usefulness of marrow morphology as a diagnostic tool.85 Nevertheless, marrow morphology was inexplicably given a dominant role in the latest WHO diagnostic algorithms.1,2
The need to assess 17 different histologic features was a major but unstated problem of the WHO prefibrotic PMF morphologic criteria, and one not easily translated to routine pathology practice, violating the WHO REAL paradigm.3 These criteria have now been refuted by a study demonstrating substantial interobserver variability for 16 of the 17 histologic features, a correlation of marrow cellularity with JAK2 V617F status, and no difference in clinical phenotype and prognosis among ET patients with respect to the presence or absence of a “prefibrotic myelofibrosis” histology.86
The conclusion from these observations together with what is known clinically about the 3 MPD is clear: PV, ET, and PMF are not static illnesses but evolve over time, and their marrow morphology will reflect the particular stage in their evolution. Because the clonal burden in these disorders expands with time and is driven by JAK2 V617F in most PV patients and approximately 50% of ET and PMF patients, MPD marrow morphology is not only a moving target but also nonspecific with respect to phenotype, in contrast to lymphomas, and acute myeloid malignancies.
Serum erythropoietin
With the discovery of erythropoietin, it was expected that the serum erythropoietin level would permit differentiation between hypoxic and autonomous erythrocytosis. Unfortunately, because erythropoietin is metabolized by its target cells87 and erythropoietin production is suppressed by erythrocytosis88 or an increase in blood viscosity,89 an increase in the red cell mass is associated with down-regulation of erythropoietin production unless hypoxia is severe.88 Thus, many patients with hypoxic erythrocytosis have a normal serum erythropoietin level.39 Although it is true that the lowest serum erythropoietin levels are found in PV,90 not only is the level often normal, but ET patients can have a similarly low serum erythropoietin level for the same hematocrit level.91 Thus, a normal serum erythropoietin level does not exclude PV as a diagnosis and a low serum erythropoietin level is not specific for it.92 Because erythrocytosis is a consequence of JAK2 V617F expression, a positive assay for this mutation in the presence of documented erythrocytosis makes the serum erythropoietin assay redundant for the diagnosis of PV.
Endogenous erythroid colony formation
The seminal discovery that PV erythroid progenitors could proliferate in vitro in the absence of erythropoietin defined the autonomous nature of this disorder.93 Although endogenous erythroid colonies (EECs) are not limited to PV,94,95 their presence in association with erythrocytosis confirms its autonomous nature.95 Unfortunately, this test was never standardized for clinical use, which is not a trivial issue with respect to its interpretation,96 and it is usually only available in research laboratories. Given the fact that EEC formation in PV is not dependent on JAK2 V617F expression,74 this assay provides no additional information; and because EEC can be observed in ET patients,97,98 the assay is not specific and should be considered a research tool of historic interest.
Alternatives to the WHO diagnostic guidelines and algorithms
Given the considerations discussed herein, we now wish to individually address the proposed WHO diagnostic guidelines and algorithms for PV, ET, and PMF and offer alternative guidelines based on physiologic principles. There are several issues central to the diagnosis of these disorders not considered by the WHO. First, given greater access to medical care and the use of electronic cell counters, MPD patients are being recognized at a younger age and earlier in the course of their disease, challenging the classic clinical and laboratory phenotypes. Second, there is no single or simple method for establishing the diagnosis of an MPD because their clinical manifestations are so pleomorphic and overlapping. Therefore, the diagnostic approach must be tailored to the patient yet not be so complex to tax physician resources.
Polycythemia vera
There are 5 major considerations with respect to the diagnosis of PV: first, it is the most common of the MPD, presumably because it represents the ultimate phenotype of activating JAK2 mutations.5,80 Second, absolute erythrocytosis is the hallmark of PV, and without it the diagnosis cannot be established, nor can PV be distinguished from its companion MPD; a positive JAK2 mutation assay in this situation only proves the presence of an MPD, not necessarily PV. As a corollary, when PV is a diagnostic consideration, a red cell mass and plasma volume determination or a phlebotomy trial are desirable. Third, plasma volume expansion, even in the absence of splenomegaly, can mask the true increase in red cell mass in PV39,56,58 (Table 1). As a consequence, hemoglobin or hematocrit values alone cannot be used to establish the presence of erythrocytosis; only red cell mass and plasma volume determinations can. Fourth, the presentation of PV is sufficiently pleomorphic that all laboratory clues need to be used (Table 2). Finally, not mentioned by the WHO but implicit in any evidence-based approach is the need in a given practice situation to define pretest probabilities with respect to the frequency with which different forms of erythrocytosis or apparent erythrocytosis are encountered. For example, in one study, idiopathic erythrocytosis and secondary erythrocytosis were 4 to 7 times more common than PV.99
The presenting blood counts and prevalence of palpable splenomegaly in 2 series of polycythemia vera patients at the time of diagnosis
| Polycythemia Vera Study Group, %100 | Malmo, %16 | |
|---|---|---|
| Erythrocytosis alone | 0 | 17 |
| Erythrocytosis and | ||
| Leukocytosis | 13 | 29 |
| Thrombocytosis | 30 | 16 |
| Leukocytosis and thrombocytosis | 57 | 38 |
| Splenomegaly (palpable) | 70 | 58 |
| Splenomegaly and | ||
| Leukocytosis | ND | 66 |
| Thrombocytosis | ND | 54 |
| Polycythemia Vera Study Group, %100 | Malmo, %16 | |
|---|---|---|
| Erythrocytosis alone | 0 | 17 |
| Erythrocytosis and | ||
| Leukocytosis | 13 | 29 |
| Thrombocytosis | 30 | 16 |
| Leukocytosis and thrombocytosis | 57 | 38 |
| Splenomegaly (palpable) | 70 | 58 |
| Splenomegaly and | ||
| Leukocytosis | ND | 66 |
| Thrombocytosis | ND | 54 |
ND indicates not determined.
Table 2 lists the presenting blood counts and prevalence of splenomegaly from 2 older series16,100 documenting the phenotypic diversity of PV. Any combination of a high red cell count and leukocytosis, thrombocytosis, or splenomegaly establishes the diagnosis. In the absence of a high red cell count, red cell mass and plasma volume studies are required. Surprisingly and inexplicably, the WHO omits blood counts and splenomegaly from its PV diagnostic criteria.
Figure 5 illustrates a diagnostic algorithm for patients who present without leukocytosis, thrombocytosis, or palpable splenomegaly, which is also useful in situations of plasma volume contraction and when the hematocrit appears to be normal because these are the patients who trouble us most and for whom the WHO provides no guidance. Bone marrow aspiration and biopsy are not required, and a JAK2 mutation assay, although desirable, is also not mandatory.

A diagnostic algorithm for suspected erythrocytosis when leukocytosis, thrombocytosis, or splenomegaly is not present. Given the likelihood that an isolated hematocrit or hemoglobin elevation will not be resulting from PV,99 the algorithm makes no a priori assumptions about the etiology of the abnormality.
A diagnostic algorithm for suspected erythrocytosis when leukocytosis, thrombocytosis, or splenomegaly is not present. Given the likelihood that an isolated hematocrit or hemoglobin elevation will not be resulting from PV,99 the algorithm makes no a priori assumptions about the etiology of the abnormality.
Finally, we take exception to the WHO contention that missing occult PV is clinically insignificant, particularly when there is no excuse for doing so. Contrary to the WHO claim, the treatment of “high-risk” MPD patients is not the same regardless of diagnosis because there is as yet no drug therapy proven to prevent venous thrombosis in PV101-104 (Figure 3), whereas phlebotomy to a sex-specific hematocrit105 rapidly alleviates the hyperviscosity symptoms unique to PV, as well as reduces the red cell contribution to thrombosis by lowering blood viscosity46 and diminishing red cell nitric oxide scavenging.106 Moreover, It would be inappropriate to treat an ET patient for PV; the assumption of PV when that disease was not present has been associated with the iatrogenic induction of acute leukemia.107,108
Essential thrombocytosis
ET is the only MPD without a specific phenotype. Because isolated thrombocytosis can be the initial clinical manifestation of PV, PMF, or chronic myelogenous leukemia, ET is not only a diagnosis of exclusion, it should also not be considered a single disease entity.109 The degree of female predominance in ET is also unique among the MPDs because of its natural history, which is compatible with a normal lifespan.110,111 The JAK2 V617F mutation provides an opportunity to identify approximately 50% of patients with isolated thrombocytosis as possibly having ET and, importantly, also further distinguishes them as possibly having PV.41,79,80 Because arterial thrombosis is more common in ET whereas venous thrombosis is more common in PV,112 this is not a trivial issue.
In addition, it is also important to consider the pretest probabilities with respect to the frequency with which clonal forms of thrombocytosis are encountered. For example, in one 4-year survey of 732 patients with a platelet count greater than 500 × 109/L (500 000/μL), only 89 (12.3%) had a clonal disorder; and of these, only 40 (5%) had ET.113 As a corollary, although the height of the platelet count is usually not diagnostic for clonally derived thrombocytosis, the higher the platelet count, the greater the degree of diagnostic specificity.114
What also appears to be important is not the chosen threshold platelet count but whether the thrombocytosis is persistent. In an epidemiologic study of 99 patients with platelet counts greater than 400 × 109/L (400 000/μL), only 8 still had thrombocytosis 8 months later.99 Thus, prior blood counts, if available, can provide valuable information. Finally, context is also important. Although ET patients can have leukocytosis and splenomegaly, if these are not extremely modest and if extramedullary hematopoiesis is present in the form of a leukoerythroblastic reaction, another MPD should be considered because these findings suggest a clonal burden larger than that usually encountered in ET (Figure 4).
Considering all these facts together, we offer the following ET diagnostic guideline (Table 3) from the perspective that, because the WHO diagnostic criteria for PV are unsatisfactory and because exclusion of PV is mandatory in the differential diagnosis of ET, the WHO PV criteria corrupt the other MPD diagnostic guidelines.
Diagnostic criteria for essential thrombocytosis
| Persistent thrombocytosis more than 400 000/μL in the absence of a reactive cause* |
| Absence of iron deficiency (normal serum ferritin for sex) |
| JAK2 V617F assay (peripheral blood; expression establishes the presence of an MPD but not its type; absence does not exclude an MPD) |
| Hemoglobin less than 16 g/dL in a man or less than 14 g/dL in a woman (hematocrit < 47% in a man or < 44% in a woman) in the absence of splenomegaly; otherwise, red cell mass and plasma volume determinations are mandatory if a JAK2 V617F assay is positive |
| Negative Bcr-Abl FISH (peripheral blood) if a JAK2 V617F assay is negative |
| If there is anemia, macrocytosis, or leukopenia, or evidence of extramedullary hematopoiesis (ie, circulating nucleated erythrocytes, immature myelocytes, or splenomegaly), a bone marrow examination (including flow cytometry and cytogenetics) is mandatory regardless of JAK2 V617F expression status |
| Persistent thrombocytosis more than 400 000/μL in the absence of a reactive cause* |
| Absence of iron deficiency (normal serum ferritin for sex) |
| JAK2 V617F assay (peripheral blood; expression establishes the presence of an MPD but not its type; absence does not exclude an MPD) |
| Hemoglobin less than 16 g/dL in a man or less than 14 g/dL in a woman (hematocrit < 47% in a man or < 44% in a woman) in the absence of splenomegaly; otherwise, red cell mass and plasma volume determinations are mandatory if a JAK2 V617F assay is positive |
| Negative Bcr-Abl FISH (peripheral blood) if a JAK2 V617F assay is negative |
| If there is anemia, macrocytosis, or leukopenia, or evidence of extramedullary hematopoiesis (ie, circulating nucleated erythrocytes, immature myelocytes, or splenomegaly), a bone marrow examination (including flow cytometry and cytogenetics) is mandatory regardless of JAK2 V617F expression status |
MPD indicates myeloproliferative disorder; and FISH, fluorescent in situ hybridization.
As indicated in the text, MPD patients represent only a minority of thrombocytosis patients in general but constitute most of those with persistent thrombocytosis in the absence of a definable cause.
Primary myelofibrosis
The recent change in nomenclature from idiopathic myelofibrosis to primary myelofibrosis115 illustrates the lack of exactness with which the MPDs are considered. Biologically, there is no such thing as “primary” myelofibrosis. Marrow fibrosis is reactive and reversible if the underlying cause can be eradicated,116 and not a disease per se. PMF is the least common of the MPDs117 and the hardest to define because of its phenotypic mimicry of a wide variety of hematologic and nonhematologic illnesses and the particular burden marrow fibrosis imposes on histologic evaluation.
PMF is the one MPD for which a bone marrow biopsy is essential for diagnostic purposes, but it is important to remember that the presence of myelofibrosis does not exclude PV.20 Although there is undoubtedly a prefibrotic phase of PMF, it is not possible at present to identify this morphologically.86 Moreover, based on most large clinical series to date (reviewed by Varki et al118 ), the absence of splenomegaly or other evidence of extramedullary hematopoiesis also makes the diagnosis of PMF suspect. As a consequence, we do not consider splenomegaly a minor criterion. This is in keeping with the high JAK2 V617F clonal burden in PMF at the time of diagnosis (Figure 4). Table 4 lists an alternative diagnostic approach for PMF.
Diagnostic criteria for primary myelofibrosis
| Leukoerythroblastic blood picture |
| Increased marrow reticulin in the absence of an infiltrative or granulomatous process |
| Splenomegaly |
| JAK2 V617F assay (peripheral blood; expression establishes the presence of an MPD but not its type; PV is always a consideration; absence does not exclude an MPD) |
| Increased circulating CD34+ cells (> 15 × 106/L) and no increase in marrow CD34+ cells by in situ immunohistochemistry |
| Characteristic cytogenetic abnormalities (peripheral blood: del(13q), 9p, del(20q), del(12p), partial trisomy1q, trisomy 8, and trisomy 9) |
| Absence of Bcr-Abl, AML, or MDS cytogenetic abnormalities by FISH (peripheral blood) |
| Leukoerythroblastic blood picture |
| Increased marrow reticulin in the absence of an infiltrative or granulomatous process |
| Splenomegaly |
| JAK2 V617F assay (peripheral blood; expression establishes the presence of an MPD but not its type; PV is always a consideration; absence does not exclude an MPD) |
| Increased circulating CD34+ cells (> 15 × 106/L) and no increase in marrow CD34+ cells by in situ immunohistochemistry |
| Characteristic cytogenetic abnormalities (peripheral blood: del(13q), 9p, del(20q), del(12p), partial trisomy1q, trisomy 8, and trisomy 9) |
| Absence of Bcr-Abl, AML, or MDS cytogenetic abnormalities by FISH (peripheral blood) |
Conclusions
JAK2 V617F expression is responsible for the most prominent clinical features of the MPDs, but its impact on disease phenotype and disease progression is clearly dependent on the total allelic burden of the mutation76,78 and other genetic influences, of which sex of the patient is a major contributor.119 Because clonal dominance is time-dependent,37 the MPDs are not static disorders and their clinical phenotype is subject to change over time. As a consequence, no single diagnostic test and, frequently, no combination of diagnostic tests, is sufficient to establish the diagnosis of a particular MPD or even to distinguish the MPDs from the other benign and malignant hematologic disorders they mimic. MPD diagnosis is still a clinical exercise; and, in this regard, it is important to remember that the “apparent absence of evidence is not evidence for its absence.” It is for these reasons that we advocate the time-tested diagnostic principles described in this review.
Acknowledgment
The authors thank Dr Alison R. Moliterno for permission to present the patient illustrated in Figure 1B.
Authorship
Contribution: J.L.S. and R.T.S. drafted and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jerry L. Spivak, Johns Hopkins University School of Medicine, Traylor 924, 720 Rutland Avenue, Baltimore, MD 21205; e-mail: jlspivak@jhmi.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal