

Abstract
Tightly associated factor Va (FVa) and factor Xa (FXa) serve as the essential prothrombin-activating complex that assembles on phosphatidylserine (PS)–containing platelet membranes during blood coagulation. We have previously shown that (1) a soluble form of PS (C6PS) triggers assembly of a fully active FVa-FXa complex in solution and (2) that 2 molecules of C6PS bind to FVa light chain with one occupying a site in the C2 domain. We expressed human factor Va (rFVa) with mutations in either the C1 domain (Y1956,L1957)A, the C2 domain (W2063,W2064)A, or both C domains (Y1956,L1957,W2063,W2064)A. Mutations in the C1 and C1-C2 domains of rFVa reduced the rate of activation of prothrombin to thrombin by FXa in the presence of 400 μM C6PS by 14 000- to 15 000-fold relative to either wild-type or C2 mutant factor rFVa. The Kd's of FXa binding with rFVa (wild-type, C2 mutant, C1 mutant, and C1-C2 mutant) were 3, 4, 564, and 624 nM, respectively. Equilibrium dialysis experiments detected binding of 4, 3, and 2 molecules of C6PS to wild-type rFVa, C1-mutated, and C1,C2-mutated rFVa, respectively. Because FVa heavy chain binds 2 molecules of C6PS, we conclude that both C2 and C1 domains bind one C6PS, with binding to the C1 domain regulating prothrombinase complex assembly.
Introduction
Activated coagulation factor V (FVa) is a protein cofactor that binds to membranes containing phosphatidylserine (PS) and regulates the production of thrombin by the prothrombinase complex.1,2 The prothrombinase complex consists of FVa, the serine protease factor Xa (FXa), calcium, and a phospholipid-containing membrane. Factor V exists in plasma as an inactive, large, single-chain glycoprotein with little or no intrinsic procoagulant activity.3,4 It is composed of repeating domains: 2 A domains (A1 and A2) at the amino-terminal end, a heavily glycosylated connecting B domain, a third A domain (A3), and 2 carboxyl-terminal C domains (C1 and C2).1,5,6 Limiting proteolysis of factor V by thrombin3,7 or FXa8 removes the B domain-producing FVa that has procoagulant activity. FVa is a heterodimer9 composed of a heavy chain (FVa-HC; A1A2; molecular weight = 105,000) and a light chain (FVa-LC; A3C1 C2; molecular weight = 74 000 or 71 000).3,7,9 Heterogeneity of the light chain is seen in both bovine and human FVa3,7,10 and is due in human FVa to alternative glycosylation at Asn2181 in the C2 domain11,12 (Figure 1). The glycosylated form is termed FVa1, whereas the other form lacking glycosylation at this site is FVa2. Human FVa2 differs from FVa1 in its ability to bind tightly to FXa in the presence of PS-containing membranes or a short-chain PS (C6PS).13-15 Human plasma FVa2 and FXa form a fully active prothrombin-activating complex in the presence of soluble C6PS.16

Amino acid residues in human FVa that contribute to PS binding. The structure of bovine FVai solved by Adams et al38 is shown as rendered by the Cn3D software package.51 Selected amino acids are labeled using the corresponding residues in human FVa. Amino acid residues previously implicated in FVa binding to PS membranes are highlighted in yellow. The indole side chains of Trp2063 and Trp2064, located in the FVa C2 domain, penetrate the lipid bilayer and contribute a majority of the free energy associated with FVa binding to PS membranes.21 The hydrophobic side chains of Tyr1956 and Leu1957 are located in a structurally analogous position within the FVa C1 domain and provide a modest contribution to the free energy associated with FVa binding to PS membranes.23 Glycosylation of Asn2181 (green) results in the FVa1 glycoform. FVa2 is not glycosylated at Asn2181.
Amino acid residues in human FVa that contribute to PS binding. The structure of bovine FVai solved by Adams et al38 is shown as rendered by the Cn3D software package.51 Selected amino acids are labeled using the corresponding residues in human FVa. Amino acid residues previously implicated in FVa binding to PS membranes are highlighted in yellow. The indole side chains of Trp2063 and Trp2064, located in the FVa C2 domain, penetrate the lipid bilayer and contribute a majority of the free energy associated with FVa binding to PS membranes.21 The hydrophobic side chains of Tyr1956 and Leu1957 are located in a structurally analogous position within the FVa C1 domain and provide a modest contribution to the free energy associated with FVa binding to PS membranes.23 Glycosylation of Asn2181 (green) results in the FVa1 glycoform. FVa2 is not glycosylated at Asn2181.
The carboxy-terminal C2 domain of FVa (residues 2037-2196 in human FVa) contains a soluble PS (C6PS) binding pocket flanked by a pair of tryptophan residues, Trp2063/Trp206417 (Figure 1). Mutating these tryptophan residues to alanine (C2 mutant) yielded FVa with impaired ability to interact with the phospholipid membrane18,19 and results in a more than 400-fold reduction in FVa2-membrane–binding affinity but does not block the assembly and activity of the prothrombinase in the presence of 25% PS-containing membranes20 or soluble C6PS.21 The binding site in the C2 domain is needed for binding of FVa to PS-containing membranes,20,21 accounts for one of 2 C6PS sites in FVa light chain,22 and is not specific for soluble C6PS,17 even though the cofactor activity of FVa2 is specifically enhanced 15 000-fold by C6PS.21 These observations suggested that a C6PS-binding site exists somewhere else in FVa light chain and might serve as a PS-specific regulatory site for assembly or activity of the rFVa2·FXa complex.21 Recently, we showed that prothrombin activation in the presence of the FVa2 mutant (Y1956,L1957)A (C1 mutant) was markedly impaired on phospholipid vesicles containing 10% or less PS but was essentially normal on vesicles containing 25% PS.23 This observation suggests that these residues in the C1 domain may somehow be involved in an interaction with PS that could be important for FVa2 cofactor activity. The 2 solvent exposed amino acids (Y1956,L1957) in the C1 domain are located analogously to the critical Trp residues in the C2 domain (Figure 1).
This work aims to test the hypothesis that the PS-regulatory site in FVa2 involves these residues and is thus located in the C1 domain analogously to the C6PS-binding site in the C2 domain. Specifically, we asked whether (Y1956,L1957)A mutant rFVa2 (C1 mutant) affects the cofactor activity of FVa2 in the presence of C6PS and the assembly of the FXa-FVa2 complex in the presence of C6PS. Finally, we determined the stoichiometries of binding of C6PS to the C1 mutant and C1-C2 mutant to confirm that a single C6PS site in the C1 domain requiring the Y1956 and L1957 residues is indeed the PS-binding site responsible for regulating the cofactor activity of FVa.
Methods
Materials
1,2-Dicaproyl-sn-glycero-3-phospho-L-serine (C6PS), 1,2-dicaproyl-sn-glycero-3-phosphocholine (C6PC), and L-serine were purchased from Avanti Polar Lipids (Alabaster, AL). Radioactive serine was purchased from American Radiolabeled Chemicals (St Louis, MO). Dansylarginine-N-(3-ethyl-1,5-pentanediyl) amide (DAPA) was obtained from Haematologic Technologies (Essex Junction, VT). [5-(Dimethylamino)-1-naphthalenesulfonyl] glutamylglycylarginyl chloromethyl ketone1 was purchased from Calbiochem (San Diego, CA). All other chemicals were ACS reagent grade or the best available grade.
Phospholipids
C6PS stocks were prepared from measured quantities of 2.5 mg/mL stock solutions in chloroform21 and used within 1 day of preparation. Radiolabeled C6PS was prepared enzymatically using phospholipase D, generously provided by Avanti Polar Lipids, radiolabeled serine (serine, L-[1-14C], HOCH2CH(NH2)COOH, specific activity 50 to 60 mCi/mmol 1.85-2.22 GBq/mmol), and C6PC. Radioactive C6PS was purified from the reaction mixture by silica gel column chromatography according to the methods as described by Comfurius and Zwaal24 with minor modifications.
Recombinant proteins
Wild-type and mutant recombinant human factor Va (rFVa) were expressed in COS cells using a B domain–deleted FV construct and purified as previously described.11,23 Activated rFV (rFVa) was prepared by incubating rFV (1.6 mg) for 30 minutes with thrombin (17 μg). After activation, 200 nM D-Phe-Pro-Arg- chloromethyl ketone was added to inactivate the thrombin. The rFVa was then purified on a Mono S HR 5/5 ion-exchange column (GE Healthcare, Piscataway, NJ) to separate factor FVa1 and factor FVa2 as described previously.11,15 We have used rFVa2 in the work described here because it is a better cofactor than rFVa1 and because only rFVa2 interacts tightly with FXa in solution with C6PS.14 The C1 mutant [(Y1956,L1957)A] and C2 mutant [(W2063,W2064)A] were expressed in COS-7 cells and purified as described.20,23 The C1-C2 mutant [(Y1956,L1957,W2063,W2064)A] purification is also described elsewhere.25
To screen 42 rFVa mutants for cofactor activity in the presence of C6PS, mutants were purified using a 1-mL High-Trap SP HP column. These preparations contained approximately 70% rFVa2 and approximately 30% rFVa1 based on sodium dodecyl sulfate–polyacrylamide gel electrophoresis20 (data not shown).
Human plasma proteins
Human factor Xa and human prothrombin and thrombin were obtained from Haematologic Technologies.
Interaction between rFVa2 and DEGR-FXa
Human DEGR FXa was prepared as described.14,26 DEGR-FXa was then dialyzed against 50 mM Tris, 0.15 M NaCl, pH 7.5, to remove free reagent.27 Titration of DEGR-FXa by rFVa2 was carried out with 1 nM DEGR-FXa (buffer: 400 μM C6PS, 20 mM Tris, 150 mM NaCl, 5 mM CaCl2, pH 7.5), with 4-minute stirred equilibration between rFVa2 additions. Several intensity readings were averaged after each addition and corrected, via controls, for dilution, buffer background, and any small amount of photobleaching, which was largely eliminated by closing the excitation slit between measurements.
Binding of C6PS to human rFVa2
Changes in the intrinsic fluorescence intensity of rFVa2 in response to added C6PS were recorded with an SLM 48 000-MHF spectrophotometer (SLM Aminco, Urbana, IL) as described previously.17,21 Key controls for the occurrence of C6PS micelles were performed by measuring the changes in pyrene fluorescence.28
The response of native or mutant rFVa2 intrinsic fluorescence to soluble C6PS was fitted to a single binding site model supplied with SigmaPlot (version 6.0; SPSS, Chicago, IL).
Effect of C6PS on prothrombin activation
Lipid-dependent cofactor activity
The lipid-induced enhancement of cofactor activity (E) of all the 42 C1 and C2 mutants was determined as described by Zhai et al22 using the following equation:
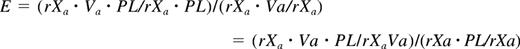
Here, rXa × Va × PL is the rate of prothrombin activation by enzyme in the presence of FVa and lipid, as measured at 400 μM C6PS as described.14,16 Other terms are defined analogously. The quantity on the left is the ratio used experimentally to evaluate E. The ratio rXa·Va/rXa was determined as the average from 5 independent experiments, whereas rXa· rXa·Va·PL/rXa·PL was the average of 3 independent measurements. The ratio on the left reflects the meaning of this quantity, as the ratio in the numerator reflects the effect of C6PS on the FXa-FVa complex, whereas that in the denominator corrects for the effect of lipid on FXa.
Determination of the stoichiometry of C6PS binding to wild-type and mutants rFVa2
The stoichiometry of soluble C6PS binding to wild-type, C1-mutated, C2-mutated, and C1-C2–mutated rFVa2 in the presence of 5 mM Ca2+ was determined by equilibrium dialysis as described previously,17,21,22 but with the modification of using radiolabeled C6PS to obtain accurate differences in C6PS concentrations between chambers using the low concentrations of recombinant rFVa2 that we had available. Experiments were performed at different rFVa2 concentrations and a fixed concentration of C6PS (0.8 and 1 μM for wild-type at 70 μM C6PS, 2 and 1.6 μM for the C1 and C2 mutants, and 2 and 2.5 μM for the C1-C2 mutant at 100 μM C6PS). These conditions ensured that at least 80% of the protein was bound to C6PS.
Results
Functional significance of binding of C6PS to the C1 domain of rFVa2
Table 1 gives the cofactor activity (determined as described in the equation in “Lipid-dependent cofactor activity”) of the wild-type and mutant rFVa2. The results clearly show that the prothrombinase activity of C2-mutated rFVa2 is the same as wild-type, as reported previously,21 whereas that of C1 mutant and the C1-C2 mutant were both reduced by approximately 14 000- and 15 000-fold, respectively, relative to wild-type rFVa2.
Summary of results with wild-type and mutant human rFVa2 and C6PS
| Proteins* | Kd, C6PS, μM | Kd, FXa, nM | Stoichiometry of binding to C6PS, no. | Cofactor activity,‡ nM/sec |
|---|---|---|---|---|
| rFVa2 | 20 ± 121 | 3.1 ± 0.4 | 4.1 ± 0.1 | 15 000 ± 455 (3) |
| rFVa2 (W2063,W2064)A | 37 ± 121 | 4.0 ± 0.6 | 3.1 ± 0.2 | 14 300 ± 470 (3) |
| rFV C2 | 2.8 ± 0.321 | NA | 1.1 ± 0.217 | NA |
| rFV C2 (W2063,W2064)A | >300† | NA | <0.0617 | NA |
| rFVa2 (Y1956,L1957)A | 47 ± 5 (3) | 564 ± 35 (3) | 3.04 ± 0.1 | 1.1 ± 0.4 (3) |
| rFVa2 (Y1956,L1957,W2063,W2064)A | NM | 624 ± 40 (3) | 2.1 ± 0.2 | 1.0 ± 0.2 (3) |
| Proteins* | Kd, C6PS, μM | Kd, FXa, nM | Stoichiometry of binding to C6PS, no. | Cofactor activity,‡ nM/sec |
|---|---|---|---|---|
| rFVa2 | 20 ± 121 | 3.1 ± 0.4 | 4.1 ± 0.1 | 15 000 ± 455 (3) |
| rFVa2 (W2063,W2064)A | 37 ± 121 | 4.0 ± 0.6 | 3.1 ± 0.2 | 14 300 ± 470 (3) |
| rFV C2 | 2.8 ± 0.321 | NA | 1.1 ± 0.217 | NA |
| rFV C2 (W2063,W2064)A | >300† | NA | <0.0617 | NA |
| rFVa2 (Y1956,L1957)A | 47 ± 5 (3) | 564 ± 35 (3) | 3.04 ± 0.1 | 1.1 ± 0.4 (3) |
| rFVa2 (Y1956,L1957,W2063,W2064)A | NM | 624 ± 40 (3) | 2.1 ± 0.2 | 1.0 ± 0.2 (3) |
Two or (where indicated in parentheses) three determinations in the present study were made. The average standard error from the nonlinear regression analysis is also given (as ±).
NM indicates not measured (not performed because the two light chain sites have been eliminated and binding of C6PS to the heavy chain of bovine FVa has previously been characterized [data fit by two linked site model: Kd1 ∼2.2 and Kd2 ∼260 μM22 ]); NA, not applicable (not applicable because C2 domain has no activity and its binding to FXa is not the subject of this study).
Kd of C1 (Y1956,L1957)A and C1C2 (Y1956,L1957,W2063,W2064)A mutants are similar to the Kd of FVa:FXa in absence of C6PS.
Kd estimated from sensitivity of the stoichiometry measurement.
The initial rates of prothrombin activation in the presence of wild-type rFVa2, the C2 mutant, the C1 mutant, and the C1-C2 mutant are plotted vs C6PS concentration in Figure 2. The data for wild-type rFVa2 (red circles) and the C2 mutant (green circles) were fitted to hyperbolas as befitting a single-site binding model. As described in Figure 2, the Kdapp's for these fits were 3.8 (± 0.5 [SEM]) μM and 4.1 (± 0.5) μM for wild-type and C2 mutant rFVa2, respectively. Previously reported values of Kdapp for bovine plasma-derived FVa (9 μM22 ) and FVa1 (11 μM) and FVa2 (10 μM) to C6PS,14 and for human rFVa2 (20 μM) determined by intrinsic fluorescence21 are somewhat larger when measured by intrinsic fluorescence changes. We note that these fluorescently determined Kdapp's reflect binding to multiple sites, 2 presumably nonphysiologic sites in the heavy chain and 2 in the light chain.22 The activity-derived Kdapp's reflect binding to whatever site(s) is (are) responsible for C6PS-triggered cofactor activity. These results demonstrate that this (these) site(s) is (are) in the C1 domain and demonstrate a Kdapp of approximately 4 μM.

Prothrombin activation in the presence of C6PS. Initial rates of human prothrombin activation were determined in reaction mixtures containing 50 mM Tris, pH 7.4, 150 mM NaCl, 5 mM CaCl2, 1 nM human FXa, 5 nM human rFVa2, 1 μM prothrombin, and different concentrations of C6PS as described in “Methods.” The rate of prothrombin activation was determined in the presence of 0 to 400 μM C6PS and rFVa2 having either an intact (A, circles) or a mutant 23C1 domain (A,B, triangles). Data obtained with rFVa2 having an intact C2 domain are shown in red (wild-type, circles in A; C1 mutant, triangles in panel B), whereas data with a mutant C2 domain18 are shown in green (C2 mutants, circles in A; and C1-C2 mutant, triangles in panel B). The lines drawn through the data in panel A are hyperbola, corresponding to a simple, single-site binding model, with parameters Kdapp = 3.8 (± 0.5) μM and saturating activity 179 (± 7) nM/sec (wild-type) and Kdapp = 3.7 (± 0.8) μM and saturating activity 177 (± 12) nM/sec (C2 mutant). Hyperbolic fits to the data up to 50 μM of C6PS are shown as dashed lines in the inset to panel A, with one of these fits (wild-type) also plotted as a dashed line in panel A. The data for rFVa2 with a mutated C1 domain were also well described by hyperbola (B), but with Kdapp's = 60 (± 4) μM and 56 (± 3) μM and saturating activities of 0.0079 (± 0.0003) and 0.0078 (± 0.0002) nM IIa/sec for the C1 and C1-C2 mutants, respectively.
Prothrombin activation in the presence of C6PS. Initial rates of human prothrombin activation were determined in reaction mixtures containing 50 mM Tris, pH 7.4, 150 mM NaCl, 5 mM CaCl2, 1 nM human FXa, 5 nM human rFVa2, 1 μM prothrombin, and different concentrations of C6PS as described in “Methods.” The rate of prothrombin activation was determined in the presence of 0 to 400 μM C6PS and rFVa2 having either an intact (A, circles) or a mutant 23C1 domain (A,B, triangles). Data obtained with rFVa2 having an intact C2 domain are shown in red (wild-type, circles in A; C1 mutant, triangles in panel B), whereas data with a mutant C2 domain18 are shown in green (C2 mutants, circles in A; and C1-C2 mutant, triangles in panel B). The lines drawn through the data in panel A are hyperbola, corresponding to a simple, single-site binding model, with parameters Kdapp = 3.8 (± 0.5) μM and saturating activity 179 (± 7) nM/sec (wild-type) and Kdapp = 3.7 (± 0.8) μM and saturating activity 177 (± 12) nM/sec (C2 mutant). Hyperbolic fits to the data up to 50 μM of C6PS are shown as dashed lines in the inset to panel A, with one of these fits (wild-type) also plotted as a dashed line in panel A. The data for rFVa2 with a mutated C1 domain were also well described by hyperbola (B), but with Kdapp's = 60 (± 4) μM and 56 (± 3) μM and saturating activities of 0.0079 (± 0.0003) and 0.0078 (± 0.0002) nM IIa/sec for the C1 and C1-C2 mutants, respectively.
Two aspects of our analysis of these data indicate that C6PS binding to rFVa2 alone triggers assembly of a prothrombinase complex with nearly full activity. First, if we fit the data from 0 to 30, 40, 50, or 400 μM C6PS (Figure 2A, inset, up to 50 μM C6PS), the best-fit parameters did not change appreciably for either the wild-type (Kdapp from 3.64-3.8 ± 0.5 μM and saturating activity from 176 to 179 ± 10 nM/sec) or C2 mutant (Kdapp from 4.0 to 4.1 ± 0.5 μM and saturating activity from 172 to 174 ± 8 nM/sec) rFVa2. Because the effect of C6PS on the activity of FXa does not saturate until approximately 300 μM C6PS (Kdapp of 70-90 μM), the steep rise in FXa activity (as seen in Figure 2) on adding C6PS must be because of lipid binding to a regulatory site on rFVa2 having a Kdapp of approximately 4 μM. This means that the dramatic effect of C6PS on the rate of prothrombin activation must be the result primarily of its binding to a regulatory site on FVa2 to trigger high-affinity binding to FXa to produce a nearly fully active prothrombinase complex. This demonstrates that the large C6PS-induced cofactor activity recorded in Table 1 is overwhelmingly the result of the ability of C6PS to produce a FVa2 conformation that binds with very high-affinity to FXa. Second, the saturating activities for both the wild-type (179 ± 7 nM/sec) and C2 mutant (177 ± 12 nM/sec) rFVa2 correspond to a kcat/KM of approximately 2.3 (± 0.1) × 108 M−1s−1, assuming a KM of 0.82 μM for the FVa2-FXa complex (average of 0.65 μM for C6PS16 or to 1 μM for membrane-assembled29 ). We have previously reported a kcat/KM of approximately 2.2 × 108 M−1s−1 for plasma-derived human FVa2-Xa complex.16 Consistent with this, the dashed curve in Figure 2A shows clearly that fitting the wild-type data from only 0 to 50 μM of C6PS underestimates only very slightly the activity at high C6PS activity. We note here and have noted previously16 that the activity of the C6PS-rVa2-FXa-C6PS complex expressed as a kcat/KM (2.2 × 108 M−1s−1) is surprisingly roughly an order of magnitude greater than 1 × 107 M−1s−129 and others30 have reported for the membrane-assembled prothrombinase obtained from a natural mixture of FVa1 and FVa2 (∼107 M−1s−1). This observation is reproducible, and the reasons for it are under investigation.
Figure 2B shows the activity of the samples containing the C1 (red triangles) and C1-C2 mutants (green triangles) in response to increasing C6PS concentration. The lines through these data were also obtained using a single site binding model with Kdapp's = 60 (± 4) μM and 56 (± 3) μM and saturating activities of 0.0079 (± 0.0003) nM IIa/s and 0.0078 (± 0.0002) nM IIa/s, respectively. The effect of C6PS on FXa alone occurs with a comparable but somewhat larger Kdapp (70 ± 10 μM31 ; 86 ± 5 μM32 ). The saturating activities (∼0.0079 and ∼0.0078 nM/sec for C1 and C1-C2 mutants) are significantly greater than previously estimated for the effect of C6PS on FXa (0.0042 nM/sec33 or 0.0036 nM/sec, R. Chattapadhyay et al, 2008, submitted for publication). This difference is probably the result of the presence of C1 and C1-C2 mutant rFVa2, which we have previously shown still enhance prothrombinase activity slightly at increasing concentrations in the presence of PS-containing membranes.23 These observations suggest that, even though C1 and C1-C2 mutant rFVa2 bind FXa very weakly, the resulting C1 and C1-C2 mutated rFVa2-FXa complexes are still more active than FXa alone.
Binding of C6PS to the C1 mutant
Because mutating the (Y1956,L1957)A residues in the C1 domain alters C6PS-stimulated prothrombin activation, it is reasonable to ask whether this mutation alters the binding of C6PS to rFVa2. Figure 3 shows the change in the intrinsic fluorescence of 0.2 μM of the C1 mutant rFVa2 resulting from titration with C6PS. The critical micelle concentration of C6PS under these conditions was determined by changes in pyrene fluorescence21 to be 700 (± 5) μM. This method is documented to detect micelles with aggregation numbers as low as 10.34 This critical micelle concentration is well above the maximum C6PS concentration used in our titration, guaranteeing that the effects reported are the result of monomeric C6PS and not micelle formation. The binding data were fit by a simple single-binding-site model assuming a stoichiometry of one (Figure 3), yielding a Kdapp of 47 (± 5) μM. Wild-type rFVa2 binds C6PS with a Kdapp of 20 (± 1) μM.21 Comparing these results, we see that elimination of the aromatic and hydrophobic side chains Tyr1956/Leu1957 from the C1 domain had little effect on the affinity of C6PS binding to rFVa2. This probably reflects the fact that FVa2 has 4 sites of roughly equivalent affinity for C6PS, so that the effect of eliminating any one would be difficult, if not impossible, to detect with a simple binding assay. Probably for the same reason, the affinity of C2-mutated rFVa2 for C6PS was also only slightly reduced (apparent Kd ∼37 μM21 ) compared with wild-type, even though we know that the C2 PS site is largely responsible for membrane binding of rFVa.20 We conclude that we cannot use simple binding affinity as a means to detect the effect of the (Y1956,L1957)A mutation on C6PS binding.

Binding of C1 mutant human rFVa2 to soluble C6PS. The intrinsic fluorescence intensities of 0.2 μM of C1 mutant rFVa2 in 50 mM Tris, 150 mM NaCl, 5 mM CaCl2, 0.6% polyethylene glycol, pH 7.5, was measured as a function of C6PS concentration at 24°C to follow C6PS binding. The data were analyzed according to a simple binding model as described in “Binding of C6PS to human rFVa2,” with results shown in Table 1. Error bars represent SEM.
Binding of C1 mutant human rFVa2 to soluble C6PS. The intrinsic fluorescence intensities of 0.2 μM of C1 mutant rFVa2 in 50 mM Tris, 150 mM NaCl, 5 mM CaCl2, 0.6% polyethylene glycol, pH 7.5, was measured as a function of C6PS concentration at 24°C to follow C6PS binding. The data were analyzed according to a simple binding model as described in “Binding of C6PS to human rFVa2,” with results shown in Table 1. Error bars represent SEM.
Stoichiometry of C6PS binding to the mutant and wild-type rFVa2
Because binding affinity could not reveal the effect of the (Tyr1956/Leu1957)A mutation on C6PS binding, we turned to direct measurement of binding stoichiometry. There are 4 C6PS-binding sites on FVa, with 2 of these residing in the light chain containing the C1 and C2 domains.22 The stoichiometries for the binding of C6PS to rFVa2, the C2 mutant, the C1 mutant, and the C1-C2 mutant were determined by equilibrium dialysis, with values given in Table 1. These results demonstrate that mutating the Tyr1956/Leu1957 residues in the C1 domain blocks binding of a second C6PS molecule to the rFVa2 light chain. We conclude that these residues are essential for expression of the second PS-binding site of rFVa2 light chain.
Binding of wild-type and mutant rFVa2 to FXa
Because the cofactor activity of FVa2 requires binding to FXa, we asked next whether C6PS binding triggers tight binding of C1-mutated rFVa2 to DEGR-labeled human FXa, as we have previously demonstrated for wild-type rFVa2 and the C2 mutant.16,21 Figure 4 presents isotherms, obtained in the presence of 400 μM C6PS, for the binding of wild-type rFVa2, C2 mutant, C1 mutant, and C1-C2 mutant of rHFVa2 to human DEGR-FXa. We have previously shown that the complex formed under these conditions with plasma-derived human FVa2 is a fully functioning prothrombin-activating complex.14,16,35 With the stoichiometry fixed at 1:1, we obtained the best fit description of the data shown in Figure 4 with Kd (and saturating F/F0 ± SEM) values of 3.1 (± 0.5) nM (1.20 ± 0.015), 4.0 (± 0.6) nM (1.42 ± 0.03), 564 (± 35) nM (1.45 ± 0.02), and 624 (± 40) nM (1.45 ± 0.02) for the wild-type rFVa2, C2 mutant, C1 mutant, and C1-C2 mutant, respectively. These results clearly show that alanine mutation of the hydrophobic (Y1956,L1957) residues of the C1 domain of rFVa2 reduces the affinity of FVa2 for FXa in the presence of 400 μM C6PS by approximately 200-fold. Note that this reduction is still less than the 1000-fold increase in affinity of FVa2 for FXa seen in the presence of C6PS,16 indicating that mutation of the Y1956,L1957 residues does not completely eliminate the effect of C6PS. This could account for the observation that FXa appears to bind C6PS with slightly more affinity in the presence of the C1 mutant rFVa2 than it does in the absence of rFVa2 (Figure 2B). We note that all 3 rFVa2 mutants produced a larger change in DEGR-FXa fluorescence than observed for the wild-type rFVa2. Although we can offer no certain interpretation of this, it is conceivable that any alterations in the C domains of FVa can be transmitted to the A2 domain that appears to be involved in interactions with FXa. Indeed, a linkage between the C1 domain and sites involved in interaction with FXa is demanded by the sum total of the results presented here.

Binding of human rFVa2 to DEGR-FXa in the presence of C6PS. Binding of factor rFVa2 to DEGR-FXa was detected by the change in fluorescence intensity of DEGR, which is covalently bound to the active site of FXa. Small aliquots of wild-type rFVa2, C2 mutant, C1 mutant, and C1-C2 mutant were added to DEGR-FXa (1 nM in 5 mM Ca2+, 50 mM Tris, 150 mM NaCl, 0.6% polyethylene glycol, 400 μM C6PS, pH 7.5). The lines through the data were obtained by least-squares regression to a simple single-site binding model with the best fit Kd values for wild-type rFVa2, C2 mutant, C1 mutant, and C1-C2 mutant being 3.1 (± 0.4) nM, 4.0 (± 0.6) nM, 564 (± 35) nM, and 624 (± 40) nM, respectively.
Binding of human rFVa2 to DEGR-FXa in the presence of C6PS. Binding of factor rFVa2 to DEGR-FXa was detected by the change in fluorescence intensity of DEGR, which is covalently bound to the active site of FXa. Small aliquots of wild-type rFVa2, C2 mutant, C1 mutant, and C1-C2 mutant were added to DEGR-FXa (1 nM in 5 mM Ca2+, 50 mM Tris, 150 mM NaCl, 0.6% polyethylene glycol, 400 μM C6PS, pH 7.5). The lines through the data were obtained by least-squares regression to a simple single-site binding model with the best fit Kd values for wild-type rFVa2, C2 mutant, C1 mutant, and C1-C2 mutant being 3.1 (± 0.4) nM, 4.0 (± 0.6) nM, 564 (± 35) nM, and 624 (± 40) nM, respectively.
Mutational analysis of the FVa C1 and C2 domains demonstrates that the amino acid side chains critical for C6PS stimulation of cofactor activity are located exclusively within the putative PS binding pocket of the C1 domain
We show here that stimulation of FVa cofactor activity by C6PS requires (Y1956,L1957); we showed previously that it is blocked by glycosylation at Asn2181, which is located in the FVa C2 domain.11,14 We asked next whether other amino acid residues within the FVa C1 and C2 domains might also contribute directly or indirectly to expression of cofactor activity in the presence of C6PS by investigating the effect of C6PS on a panel of 42 previously characterized FVa mutants18,23 (Figure 5). These results indicate that Tyr1956 and Leu1957 contribute approximately equally to the expression of FVa cofactor activity in the presence of C6PS. Several other amino acid side chains within the C1 domain appeared to make small but statistically significant contributions to the expression of cofactor activity in the presence of C6PS. These include (Lys1954/His1955), Glu1964, and Arg2023. Each of these residues is located within or in close proximity to an apparent “pocket” that is structurally (Figure 1) related to the C6PS binding pocket that we have demonstrated in the C2 domain.17 In contrast, none of the 22 C2 domain mutants had a significant effect on rFVa cofactor activity in the presence of C6PS.

Activity of human rFVa C1 and C2 domain mutants in the presence of C6PS. The cofactor activity of rFVa mutants was determined as described in the equation 1 in “Lipid-dependent cofactor activity.” Reaction mixtures contained 1 μM prothrombin, 1 nM FXa, 5 nM rFVa, 5 μM DAPA in 50 mM Tris, 150 mM NaCl, 5 mM Ca2+, pH 7.5. Shown are means (± SD) of 4 measurements. *P < .002 compared with wild-type rFVa.
Activity of human rFVa C1 and C2 domain mutants in the presence of C6PS. The cofactor activity of rFVa mutants was determined as described in the equation 1 in “Lipid-dependent cofactor activity.” Reaction mixtures contained 1 μM prothrombin, 1 nM FXa, 5 nM rFVa, 5 μM DAPA in 50 mM Tris, 150 mM NaCl, 5 mM Ca2+, pH 7.5. Shown are means (± SD) of 4 measurements. *P < .002 compared with wild-type rFVa.
Discussion
Roles of the FVa C domains in FVa function
The C domains of FVa contribute both to membrane binding and to regulating the ability of FVa to bind with high affinity to FXa, with both processes being dependent on binding of PS molecules to these domains. Support for this conclusion derives both from our previously published results and the new observations presented here on the role of PS binding to the C1 domain in regulating FVa binding to FXa.
The C2 domain
Previous work.
The conclusion that the C2 domain is primarily responsible for membrane binding rather than cofactor activity follows from several previous reports. First, the structure of the C2 domain of human FVa is a β-sandwich that caps an apparent “pocket” at one end of the β-sheet structure.36 Two partially hydrophobic loops that protrude (one in a β-loop and the other in a near β-loop) from the “bottom” end of the β-sandwich have hydrophobic residues at their tips (Figure 1). In the first loop, the hydrophobic residues are a pair of tryptophans (W2063,W2064), and in the other a leucine (L2116). These and another β-loop form a “pocket.” Second, the free energy of rFVa2 binding to 25% PS-containing membranes differs from that of a C1-mutated rFVa2 by only approximately 1 kBT.21 Third, use of a (W2063,W2064)A mutant rFVa2 and expressed C2 domain showed that the (W2063,W2064) residues are critical for both C6PS (Kd > 300 μM16 ) and tight membrane binding (Kd ≫ 100 nM16,20 ). However, these tryptophans are not needed for formation of a fully active prothrombinase complex in solution.16 They are also not required for prothrombinase activity on a 25% PS-containing membrane as long as concentrations of rFVa2 and FXa are used that are 10-fold and 4-fold higher, respectively, than required to reach maximal activity with wild-type rFVa2.20 Fourth, these Trp residues insert into the bilayer interface during membrane binding.21 Fifth, labeling of a Cys at the top of the C2 pocket blocked binding of C6PS to rFVa-C2 domain.17 Thus, the (W2063,W2064) indole side chains, which form the rim of the pocket on the C2 domain, define the PS-binding pocket and account for nearly all of the free energy of binding of rFVa2 to 25% PS-containing membranes.
The C1 domain
Previous work.
The results summarized in “The C2 domain, Previous work” suggest that another PS site on rFVa2 must account for the clear regulatory role of C6PS in triggering assembly of a fully active prothrombinase complex.21 Two C6PS sites exist in the FVa light chain and 2 in the heavy chain,22 with the heavy chain sites quite probably being of no physiologic significance because of the large distance that probably separates the heavy chain from the membrane.37 In addition, the C1 domain is similar to the C6PS-binding C2 domain. Finally, a double C218 and C123 mutant interfered with both membrane binding and appearance of an active prothrombinase complex.25 For these reasons, we undertook the current study to examine the effects of C6PS on the C1 domain.
Because the isolated human C1 domain has yet to be expressed, isolated, and crystallized, somewhat less is known about its properties than about the C2 domain. However, a recent crystal structure of APC-inactivated bovine FVa38 (Figure 1) shows that the C1 domain is structurally analogous to that of the C2 domain. It too has a partially hydrophobic β-loop that extends down from the β-sandwich, with a hydrophobic pair (Y1956,L1957) at its tip, and with structural similarity to the Leu2116 loop of the C2 domain. At least in this structure, the tip of the C1 loop is roughly in the same plane as the tips of the 2 C2 loops with the 2 C domains roughly perpendicular to this plane, so that one could imagine the entire loop tips located at the hydrophobic/hydrophilic interface of a membrane lipid bilayer (Figure 1). Two other, non-β loops help form a “pocket” in the C1 domain.
Current work.
If this x-ray-derived model has relevance to the structure of FVa on a membrane, it is probable that only the C6PS-binding sites in the light chain are physiologically important. We have previously suggested that the 2 C6PS sites located in the FVa heavy chain are anomalous and correspond to protein recognition sites.22 Here we focus on the C1 domain PS-binding sites and show by alanine mutagenesis that (1) the (Y1956,L1957)A mutation is unique in its ability to affect prothrombinase activity (Figure 5); (2) the (Y1956,L1957)A mutation eliminates the PS-triggered cofactor activity of rFVa2 (Figure 2; Table 1); (3) the (Y1956,L1957)A mutation reduces the C6PS-induced affinity of rFVa2 for FXa by approximately 200-fold (Figure 4); and (4) the (Y1956,L1957)A mutation eliminates one of 2 binding sites for C6PS in the light chain of rFVa2 (Table 1).
All these conclusions support the hypothesis that the Y1956 and L1957 residues line a PS-binding pocket that regulates FVa cofactor activity. Because we have been unable to isolate expressed rHFVa2 C1 domain, we do not have the cystine labeling data that we used to block and thus locate with certainty the PS-binding site in the C2 domain.17 However, the structural similarity between the C1 and C2 domains, along with the mutational analysis presented here, strongly supports this hypothesis.
Model for the role of C1 and C2 domains in FVa2
These observations and other previously published observations on the C2 domain16,20-23,25,33,39 suggest a model for FVa membrane binding and activation by PS, as summarized in Figure 6. In this model, binding of wild-type rFVa2 to a PS-containing membrane (top panel) occurs primarily (but not exclusively16,20 ) via the C2 domain. It is well known that extrinsic membrane proteins, including FXa and FVa, can bind to membranes through multiple interaction sites.40,41 In the case of rFVa2, the interaction that overcomes most of the unfavorable free energy of moving the protein from a 3-dimensional to 2-dimensional environment comes from the C2 domain.21 However, once rFVa2 is on the surface, the less avid C1 site can be occupied. This triggers a change in rFVa2 that causes it to bind with greatly increased affinity to FXa (Figure 4).16,22 FXa itself is activated by approximately 60-fold through binding to C6PS or to a PS-containing membrane33,39 but is optimally active when bound to FVa2.16 This results in the fully active prothrombinase.

Proposed model of prothrombinase assembly for wild-type human FVa, and for C1- and C2-mutated FVa. This model is based on the well-known fact that the free energy of binding of extrinsic membrane proteins to membranes often reflects multiple interactions, and on the results presented in this paper.
Proposed model of prothrombinase assembly for wild-type human FVa, and for C1- and C2-mutated FVa. This model is based on the well-known fact that the free energy of binding of extrinsic membrane proteins to membranes often reflects multiple interactions, and on the results presented in this paper.
The model in Figure 6 accounts for all our current and past observations on lipid effects on factors FVa and FXa. Our previous reports have established that, despite the important functions that we have demonstrated for the C2 and C1 domains of FVa, both the C1 mutant and C2 mutant forms of rFVa2 can form active prothrombin-activating complexes with FXa on membranes containing 25% PS.20,23 In terms of our model, the ability of the (W2063,W2064)A rFVa2 (C2) mutant to support formation of an active prothrombinase on 25% PS-containing membranes20 can be explained by the presence of a very small amount of mutant rFVa on a membrane (eg, Kd of FVa binding to a phosphatidylcholine membrane ∼ 0.3 μM33 ; Kd ≫ 100 nM for binding to 25% PS membranes20 ). C2 mutant FVa2 on a PS-containing membrane should still interact normally with the intact C1 domain, leading to high-affinity binding with FXa (Kd ∼0.6 nM21 ). Because FXa binds with reasonable affinity to PS-containing membranes (Kd ∼0.15 μM40 ), binding of FXa to a membrane can also enhance assembly of a C2 mutant-containing prothrombinase complex on a membrane. Mass action then will drive formation of an active prothrombinase, but at slightly higher rFVa2 or FXa concentrations than would be required with wild-type rFVa2.
The affinity of the (Y1956,L1957)A rFVa2 (C1) mutant for 25% PS-membranes is reduced only slightly compared with that of wild-type rFVa2,16,23 so it locates to the membrane surface very effectively. However, PS does not bind with normal affinity to the regulatory site in the mutated C1 domain (Table 1), meaning that it binds FXa with approximately 200-fold lower affinity than does wild-type rFVa2 (Figure 4). Nonetheless, a solution Kd for the FXa-rFVa2 complex of 0.5 to 0.6 μM (9.1-10.9 × 10−9 mol fraction) corresponds to a unitary free energy of association (ΔG°) of −18.5 to −18.3 kBT. Under the conditions used in our previous paper (0.2 nM C1-mutated rFVa, and 5 μM lipid in membranes23 ), nearly all rFVa should be membrane bound. Assuming that this same ΔG° of association holds for the membrane-associated FXa and rFVa2, we can obtain the fraction of surface-bound FXa bound to surface-bound C1 mutant rFVa2 by simply expressing surface concentrations in mol fraction units. If we do so, we find that it would take only 6 nM FXa to essentially saturate (99.9% bound) the membrane-bound C1 mutant rFVa and thereby obtain full prothrombinase activity. In solution, the same concentrations of FXa and C1 mutant rFVa2 would yield approximately 2 fM prothrombinase. Thus, the profound effect of dimensionality reduction can explain why C1 mutant rFVa2 can form a prothrombinase complex in the presence of 25% PS membranes, albeit at approximately 8- to 10-fold higher FXa concentration than is required for wild-type rFVa2.23 The fact that even higher concentrations of FXa or C1 mutant rFVa2 still do not produce full prothrombinase activity23 reflects the much weaker binding of these proteins to membranes with low PS content.40,41 Taken together, the current results along with our previous observations23 indicate that the C6PS-binding pocket defined by the (Y1956,L1957)A mutation regulates the affinity of rFVa2 for FXa but not the effect of FVa on FXa activity.
It is worth noting that the turnover number of this membrane-assembled C1 mutant rFVa2 complex is within error limits the same as that seen with wild-type rFVa2 complex.23,25 Thus, although the (Y1956,L1957)A mutation clearly affects the assembly of the prothrombinase complex,21 it does not affect significantly the activity of the prothrombinase complex once it is assembled. This suggests that the tight-FXa-binding conformation induced by PS binding to the C1 domain has little effect on cofactor activity other than being a conformation that promotes formation of the FXa-FVa complex.
The double-mutant (Y1956,L1957,W2063,W2064)A rFVa2, should not bind tightly to membranes and should not be activated to a form with high affinity for FXa. According to our model, we expect that it will have little, if any, activity in the presence of PS-containing membranes. This is what we have observed.25
Our results indicate that PS not only helps localize FVa to the membrane surface but also serves to enhance cofactor activity through enhanced FXa binding affinity. Both the C1 and C2 domains are most probably important during the initial phases of hemostasis when concentrations of FXa and exposed PS may be limiting.42 Under these conditions, the C2 domain is needed to locate FVa to a platelet membrane. In addition, conformational changes in the C1 domain resulting from the binding of PS presumably result in conformational changes elsewhere in FVa that lead to enhanced FXa binding. FXa-binding sites in FVa have been localized by peptide inhibition studies to the A1/A2 domain interface,43,44 by loop swapping to the A2 domain,45 by limited proteolysis to the A2 domain,46 and finally by site-directed mutagenesis to the A2 and A3 domains of FVa.47 The latter mutagenesis study identifies His1683 as the only A3 domain residue key to FXa binding. This is a surface-located histidine not far from the A2/A3 interface. Because all other studies identify residues in the A2 domain (or A1/A2 interface) as involved in FXa binding, it seems probable that the binding of C6PS to the C1 domain may lead to conformational changes in the adjacent A3 domain that then transfer the signal to the A2 domain to produce a high-affinity FXa binding configuration.
In conclusion, if mutations in the C1 domain that seriously inhibit binding of PS to this domain fail to block prothrombinase complexes on membranes containing 25% PS, what can be the importance of this domain physiologically? The answer lies in the fact that the platelet membrane contains only approximately 10% PS,48 and it is probable that the activated platelet vesicles that support coagulation49,50 expose only 5% to 10% PS on their surfaces. On membranes containing only 5% PS, C1 mutant rFVa is dramatically ineffective in supporting prothrombin activation.23 From this observation and the results presented here, it is probable that PS binding to the C1 domain of FVa plays a key regulatory role in blood coagulation.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank reviewers of previous versions of this manuscript for anonymous comments.
This work was supported by National Institutes of Health, Bethesda, MD (grants HL72827, B.R.L.; and HL43106, W.H.K.).
National Institutes of Health
Authorship
Contribution: R.M. and B.R.L. designed experiments; R.M. performed experiments, collected, analyzed, and interpreted data, performed statistical analysis, and wrote the initial manuscript; M.A.Q.-A. expressed, purified, and characterized the recombinant proteins; and W.H.K. and B.R.L. sponsored the research, helped interpret the data, and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Barry R. Lentz, Department of Biochemistry and Biophysics, CB#7260, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7260; e-mail: uncbrl@med.unc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal