

Abstract
The effect of induction therapy with multiple doses of rituximab on the subsequent efficacy and toxicity of anti-CD20 radioimmunotherapy is unknown. We evaluated a novel protocol using 4 weekly infusions of 375 mg/m2 rituximab followed by 2 fractions of 131I-rituximab, preceded by a 100-mg/m2 predose of rituximab, in relapsed indolent B-cell lymphoma. Induction therapy with rituximab significantly increased the effective half-life of 131I-rituximab (P = .003) and high serum levels of rituximab after induction therapy correlated with increased effective half-life of the radioimmunoconjugate (P = .009). Patients with large tumor burdens experienced significant increases in the effective half-life of 131I-rituximab between delivery of the first and second fractions (P = .007). Induction therapy with multiple doses of rituximab did not appear to compromise the clinical efficacy or increase toxicity of subsequent 131I-rituximab radioimmunotherapy. The overall response rate was 94%, with complete response rate 50%. The median time to progression was 20 months, significantly longer than for the last qualifying chemotherapy (P = .001). Fractionation of 131I-rituximab allowed cumulative whole-body doses of more than 120 cGy, approximately 60% greater than those previously achieved with a single administration of a murine radioimmunconjugate, to be delivered without significant hematologic toxicity.
Introduction
The majority of indolent non-Hodgkin lymphomas (NHLs) present with advanced-stage disease, and, despite initial sensitivity to therapy, most patients will eventually relapse and die of their disease.1 The addition of the anti-CD20 monoclonal antibody (mAb) rituximab to chemotherapy has substantially improved response rates and has improved outcomes.2-4 An alternative approach to improving response rates with anti-CD20 mAb involves the conjugation of radioisotopes to the mAbs, or radioimmunotherapy (RIT). Two radiolabeled murine anti-CD20 mAbs have been licensed by the US Food and Drug Administration (FDA), namely, 131I-labeled tositumomab (Bexxar, GlaxoSmithKline, Philadelphia, PA) and 90Y-labeled ibritumomab tiuxetan (Zevalin, Bayer Schering, Berlin, Germany). Both drugs yield impressive overall response rates (ORRs) of approximately 75% to 80% and complete response (CR) rates of 20% to 50%, with durable remissions when administered as a single nonmyeloablative fraction in relapsed follicular lymphoma.5,6
Despite these impressive clinical results when using RIT in indolent lymphomas, the optimal treatment approach remains undefined. An important area of current uncertainty in the delivery of RIT concerns whether prior therapy with rituximab compromises the efficacy and toxicity profile of subsequent RIT. The initial pivotal trials that led to regulatory approval for the approved RIT drugs were performed before the routine use of rituximab containing immuno-chemotherapy. These early studies demonstrated that predosing with “cold” or unlabeled anti-CD20 mAb before the delivery of the RIT improved the subsequent biodistribution and delivery of the radioimmunoconjugate.7 Subsequently, a number of phase 2 studies have demonstrated that using 131I-tositumomab after initial chemotherapy improves the quality of the clinical response, converting many partial responses (PRs) to CRs.8,9 Furthermore, in a recently reported randomized study, administering 90Y-labeled ibritumomab tiuxetan (Zevalin) after initial chemotherapy resulted in a 77% conversion from PR to CR, leading to a 2-year improvement in progression-free survival.10 However, none of the phase 2 studies included rituximab in the induction chemotherapy regimen, and there were only a small minority of patients in the randomized FIT study who received rituximab. Therefore, the effect that rituximab-containing immunochemotherapy may have on the subsequent delivery of RIT remains unknown. Recent data using a preclinical lymphoma xenograft model suggested that larger doses of rituximab may indeed block subsequent binding of radiolabeled anti-CD20 mAb.11 Currently, there is no evidence from clinical studies that prior rituximab may compromise subsequent anti-CD20–based RIT. Furthermore, little is known about the effect of tumor burden on the serum rituximab concentration and the relationship between these parameters and the biodistribution of anti-CD20–based RIT.
There is also controversy regarding whether a radiation dose response exists in RIT. Compelling results have been obtained using higher myeloablative radiation doses of 131I anti-CD20 with autologous stem cell support, which appear to have led to improved response rates and longer response durations over those seen with lower nonmyeloablative doses.5,6,12 Although there have been no comparative studies performed, these data are at least hypothesis generating, in that there may be a radiation dose response for RIT. We therefore hypothesized that giving more than one fraction of 131I anti-CD20 RIT would allow a higher cumulative whole-body absorbed dose (WBD) and a higher tumor dose to be delivered, without requiring bone marrow support. A potential advantage to using fractionated RIT is the possibility of improving tumor penetration by radioimmunoconjugate in patients with bulky or larger volumes of disease with a second administration. Because mAbs are large macromolecules, a single dose may not distribute homogeneously throughout initially bulky tumor masses,13,14 and a second fraction of RIT may improve the biodistribution in larger masses because of a reduction in tumor bulk after the first dose of RIT.15 Finally, the use of a chimeric mAb makes fractionation of RIT feasible as development of human antimouse and human antichimeric antibodies is rare, occurring in as few as 1% of patients.16
Here we report the results of a phase 1/2 dose escalation study designed to evaluate the feasibility, efficacy, and safety of fractionated 131I-rituximab in relapsed or refractory indolent CD20+ NHL. We have determined the effect that induction therapy with 4 weekly infusions of rituximab has on the subsequent delivery of 131I-rituximab. For the first time, serum rituximab levels have been measured by enzyme-linked immunsorbent assay (ELISA) alongside dosimetric calculation of the effective half-life of 131I-rituximab using sequential whole-body gamma camera imaging. We demonstrate that induction therapy with rituximab significantly increases the effective half-life of 131I-rituximab and that high serum levels of rituximab after induction therapy correlate with increased effective half-life of the 131I-rituximab. Importantly, we show, for the first time, that induction therapy with multiple doses of rituximab does not appear to compromise the clinical efficacy or increase the toxicity of subsequent anti-CD20–targeted RIT.
Methods
Patients and inclusion/exclusion criteria
Written informed consent was obtained from all patients in accordance with the Declaration of Helsinki. Institutional review board approval from the Southampton and South West Hampshire local ethics committees was granted in accordance with United Kingdom Medical Research Council guidelines. Eligible patients were required to have histologically confirmed relapsed or refractory CD20+ NHL (follicular, small lymphocytic, mantle cell, or transformed), to be at least 18 years old, have a World Health Organization performance status of less than 3 and a life expectancy of more than 3 months. Exclusion criteria included blood neutrophils less than 1.5 × 109/L or platelets less than 100 × 109/L; significant impairment of cardiac, renal, or hepatic function; or the administration of chemotherapy or radiotherapy within 6 weeks. The degree of bone marrow involvement was not a study exclusion criterion, but patients with marrow involvement at entry were required to have a repeat bone marrow biopsy to determine that the level of involvement was less than 25% before receiving 131I-rituximab.
Four patients were included in each cohort. In total, 18 patients were recruited to the study over a 4-year period at Southampton University Hospitals NHS Trust, and 16 completed the study protocol. Two patients were excluded before 131I-rituximab administration through failure to meet eligibility criteria with thrombocytopenia and 50% lymphomatous bone marrow involvement, respectively. Patient 13 was excluded from statistical analyses involving calculated splenic and lymph node volume, having undergone a previous splenectomy. Demographic data for the study participants are summarized in Table 1.
Demographic characteristics of study participants (n = 16)
| Characteristic | Value |
|---|---|
| Median age, y (range) | 46.5 (34-77) |
| Sex | |
| Male | 6 (38%) |
| Female | 10 (62%) |
| Serum LDH | |
| Normal | 12 (75%) |
| Elevated | 3 (19%) |
| Unknown | 1 (6%) |
| Histology | |
| Follicular | 12 (75%) |
| Transformed follicular | 3 (19%) |
| Small lymphocytic | 1 (6%) |
| Maximum tumor diameter | |
| > 50 mm | 5 (31%) |
| < 50 mm | 11 (9%) |
| No. of nodal sites > 5 | 11 (69%) |
| FLIPI index, mean score (range) | 2.75 (0-3) |
| ECOG performance status | |
| 0 | 8 |
| 1 | 7 |
| 2 | 1 |
| Bone marrow involvement | |
| > 25% | 1 (6%) |
| 11%-25% | 4 (25%) |
| 1%-10% | 5 (31%) |
| None | 6 (38%) |
| Number of previous chemotherapy regimens, median (range) | 2 (1-4) |
| Prior rituximab | |
| Yes (4/7 rituximab refractory) | 7 (44%) |
| No | 9 (56%) |
| Characteristic | Value |
|---|---|
| Median age, y (range) | 46.5 (34-77) |
| Sex | |
| Male | 6 (38%) |
| Female | 10 (62%) |
| Serum LDH | |
| Normal | 12 (75%) |
| Elevated | 3 (19%) |
| Unknown | 1 (6%) |
| Histology | |
| Follicular | 12 (75%) |
| Transformed follicular | 3 (19%) |
| Small lymphocytic | 1 (6%) |
| Maximum tumor diameter | |
| > 50 mm | 5 (31%) |
| < 50 mm | 11 (9%) |
| No. of nodal sites > 5 | 11 (69%) |
| FLIPI index, mean score (range) | 2.75 (0-3) |
| ECOG performance status | |
| 0 | 8 |
| 1 | 7 |
| 2 | 1 |
| Bone marrow involvement | |
| > 25% | 1 (6%) |
| 11%-25% | 4 (25%) |
| 1%-10% | 5 (31%) |
| None | 6 (38%) |
| Number of previous chemotherapy regimens, median (range) | 2 (1-4) |
| Prior rituximab | |
| Yes (4/7 rituximab refractory) | 7 (44%) |
| No | 9 (56%) |
ECOG indicates Eastern Cooperative Oncology Group.
Protocol schedule
All patients received 4 loading doses of 375mg/m2 unlabeled rituximab intravenously at weeks 0, 1, 2, and 3. Dosimetry with a 185-MBq tracer of 131I-rituximab was first performed at week 7. A lower predose of 100 mg/m2 was given before the dosimetric and therapeutic 131I-rituximab infusions. The first therapeutic fraction of 131I-rituximab was subsequently administered at week 9. Dosimetry was repeated at week 15, with the second and final therapeutic fraction of 131I-rituximab delivered at week 17. The dosimetry of 5 patients was assessed with a 185-MBq dosimetric tracer dose of 131I-rituximab before receiving any rituximab. The schedule is illustrated in Figure 1A. Whole-body computed tomography (CT) scanning was performed on all patients at week 0 to assess baseline splenic and lymph node volumes and at week 25 to help determine therapeutic response. In addition, a subgroup of 5 patients received a 185-MBq tracer of 131I-rituximab before the 4 loading doses of unlabeled rituximab.
Calculation of splenic and lymph node volume
Splenic and lymph node volumes were quantified after whole-body cross-sectional CT scanning performed before study entry. A specialized software package on GE Advantage Work Station 4.1 (GE Healthcare, Little Chalfont, United Kingdom) was used to calculate volumes, which were determined independently by 2 radiologists as described by Sohaib et al.17
Measurement of serum rituximab levels
Predose (trough) and postdose (peak) serum rituximab levels were quantified weekly by ELISA. A high affinity mAb (MB2A4) specific for the idiotype region of rituximab was produced by our group and validated in ELISA as previously described.18 In brief, ELISAs using this antibody detect rituximab in the presence or absence of high levels of human IgG and are sensitive to serum rituximab levels of 10 ng/mL.
Statistical analysis
Spearman rank correlation coefficient was used to determine the significance of nonparametric correlations between tumor volume, serum rituximab levels, and the effective half-life of 131I-rituximab. The paired 2-tailed Student t test was used to determine the significance of changes in effective half-lives and durations of response to therapy. The relationship between effective half-life and serum rituximab concentration was modeled around a line of best fit, which exponentially approaches an asymptotic value for effective half-life.
Preparation of 131I-rituximab
Rituximab was radioiodinated with 131I-sodium iodide (MDS Nordion, Fleurus, Belgium) using the Iodo-Beads labeling method19 (Perbio Science United Kingdom, Cramlington, United Kingdom). Briefly, 5 mg of clinical grade rituximab (Roche Pharmaceuticals, Welwyn Garden City, United Kingdom) was added to 40 to 340 μL of 131I (300 MBq to 3.0 GBq), followed immediately by the addition of 5 Iodo-Beads for trace studies and 10 to 15 Iodo-Beads for therapeutic preparations. A labeling efficiency of 95.0% to 98.4% was achieved in 70 consecutive labeling procedures with specific activities of 60 to 600 MBq/mg. High performance liquid chromatography analysis demonstrated stable and uniform postlabeling mAb quality without purification. The mean immunoreactivity of 131I-rituximab in 70 separate labeling experiments was 63.3% plus or minus 5.75% (SD), specific binding as measured on 107 Daudi cells. Within 2 to 3 hours of radiolabeling, 131I-rituximab was infused intravenously using an infusion pump and a 0.22-μm filter in the setup of the primary line. To prevent thyroid uptake of free 131I, daily administration of potassium iodate was commenced 3 days before administration of the radioimmunoconjugate and continued for a total of 35 days.
Dosimetric calculation of the effective half-life of 131I-rituximab
The administered activity required to deliver a given WBD was determined according to the method described by Wahl et al.20 After premedica-tion with acetaminophen 1 g orally and chlorpheniramine 8 mg orally, 100 mg/m2 unlabeled rituximab was administered as an intravenous infusion, followed 2 to 3 hours later by a 20-minute infusion of 185 MBq 131I-rituximab in 30 mL 0.9% saline solution. Whole-body anterior and posterior gamma camera images were obtained within 1 hour of completing the tracer infusion dose and on days 4 and 6 after 131I-rituximab administration. Quantitative whole-body gamma camera scans were performed under the imaging conditions previously described.20 The 131I activity recorded at the time of the first scan was designated as “100%,” and the percentage activity remaining at each time point was calculated from quantitative analysis of the subsequent gamma camera images. This was plotted on a semilogarithmic scale and a best-fit line was drawn. The effective half-life of 131I-rituximab was thus calculated as the time taken for whole-body activity to fall to 50%.20 A source of standard activity was also imaged as a quality control measure.
Calculation of administered 131I-rituximab activity to deliver a predetermined therapeutic WBD
The following equation was used to calculate the activity of 131I-rituximab to be administered to give a desired WBD for an individual patient:
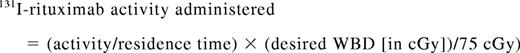
The residence time is the time taken for activity to fall to 37%, and is equivalent to 1.443 times the effective half-life, as calculated by dosimetry. The activity hours parameter is obtained from a table derived from the Medical Internal Radiation Dose system21 using the patient's height and weight and assuming that the patient is ellipsoid in shape and that the radiolabeled antibody is distributed uniformly through the patient's body after injection.22 As 131I-rituximab does not distribute into fatty tissues, the patient's maximum effective mass (related to lean body mass) was used to calculate the activity hours parameter. The patient's weight was compared with a table of effective mass, in patient's height and gender.20 If the patient's actual weight was greater than his or her maximum effective mass, the kilogram value in the table was used to calculate the therapeutic dose. If the patient's weight was less than the kilogram value in the table, the patient's actual weight was used to calculate the therapeutic dose.
Therapeutic dose escalation
Therapeutic fractions were administered at weeks 9 and 17, 2 weeks after each dosimetric study, alongside an additional 100-mg/m2 loading dose of unlabeled rituximab. The 8-week interval between fractions was chosen to allow hematologic recovery without compromising treatment efficacy. Therapeutic radiation doses were based on the dose-escalation studies of Kaminski et al with 131I-tositumomab.23 Cohort A (patients 1-4) received 2 therapeutic fractions of 131I-rituximab, each delivering 15 cGy WBD; cohort B (patients 5-8), 2 therapeutic fractions of 30 cGy each; cohort C (patients 9-12), 2 fractions of 45 cGy each; and cohort D (patients 13-16), 2 fractions of 60 cGy each. Table 2 details the total WBD delivered to each patient over the entire study schedule, including activity derived from dosimetric tracer administration.
Tumor characteristics, radiation doses, and treatment responses
| Patient no. | Histology (stage) | Radiologically calculated splenic and lymph node volume, mL | Maximum tumor diameter, mm | No. of nodal sites | FLIPI score | Previous rituximab | Previous response to chemotherapy (length in mo) | Response to 131I-rituximab (length in mo) | Cumulative whole-body dose, cGy |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Follicular (IV B) | 1320 | 60 | > 5 | 3 | Y (refractory) | PR (12) | CR (60) | 46.7 |
| 2 | Follicular (II B) | 147 | 30 | 3 | 0 | N | CR (5) | CR (60) | 43.1 |
| 3 | Transformed follicular (III A) | 290 | 30 | 4 | 1 | N | PR (10) | MR (17) | 38.4 |
| 4 | Follicular (IV A) | 1494 | 10 | > 5 | 3 | N | PR (10) | PR (23) | 41.0 |
| 5 | Follicular (IV A) | 489 | 23 | > 5 | 3 | N | PR (7) | CR (12) | 75.3 |
| 6 | Follicular (IV B) | 838 | 32 | > 5 | 2 | Y (refractory) | PR (6) | PR (13) | 72.5 |
| 7 | Follicular (IV A) | 662 | 49 | > 5 | 2 | Y | PR (4) | PR (7; transformed at relapse) | 71.1 |
| 8 | Transformed follicular (IV A) | 630 | 42 | > 5 | 3 | N | Refractory to treatment | PR (7) | 79.5 |
| 9 | Small lymphocytic (IV A) | 484 | 45 | 4 | 3 | N | CR (14) | CR (50+) | 106.5 |
| 10 | Transformed follicular (II B) | 308 | 32 | 4 | 1 | Y | PR (2) | PR (25) | 105.2 |
| 11 | Follicular (IV A) | 842 | 42 | > 5 | 2 | Y (refractory) | MR (6) | CR (45+) | 106.0 |
| 12 | Follicular (IV B) | 1451 | 73 | > 5 | 3 | Y | PR (11) | PR (25) | 95.6 |
| 13 | Follicular (IV A) | 127 | 37 | 3 | 1 | Y (refractory) | PR (3) | CR (41+) | 141.3 |
| 14 | Follicular (IV A) | 676 | 53 | > 5 | 3 | N | PR (7) | CR (28+) | 131.8 |
| 15 | Follicular (III A) | 169 | 23 | > 5 | 2 | N | PR (3) | CR (27+) | 138.72 |
| 16 | Follicular (IV B) | 679 | 58 | > 5 | 2 | N | CR (22) | PR (20) | 128.3 |
| Patient no. | Histology (stage) | Radiologically calculated splenic and lymph node volume, mL | Maximum tumor diameter, mm | No. of nodal sites | FLIPI score | Previous rituximab | Previous response to chemotherapy (length in mo) | Response to 131I-rituximab (length in mo) | Cumulative whole-body dose, cGy |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Follicular (IV B) | 1320 | 60 | > 5 | 3 | Y (refractory) | PR (12) | CR (60) | 46.7 |
| 2 | Follicular (II B) | 147 | 30 | 3 | 0 | N | CR (5) | CR (60) | 43.1 |
| 3 | Transformed follicular (III A) | 290 | 30 | 4 | 1 | N | PR (10) | MR (17) | 38.4 |
| 4 | Follicular (IV A) | 1494 | 10 | > 5 | 3 | N | PR (10) | PR (23) | 41.0 |
| 5 | Follicular (IV A) | 489 | 23 | > 5 | 3 | N | PR (7) | CR (12) | 75.3 |
| 6 | Follicular (IV B) | 838 | 32 | > 5 | 2 | Y (refractory) | PR (6) | PR (13) | 72.5 |
| 7 | Follicular (IV A) | 662 | 49 | > 5 | 2 | Y | PR (4) | PR (7; transformed at relapse) | 71.1 |
| 8 | Transformed follicular (IV A) | 630 | 42 | > 5 | 3 | N | Refractory to treatment | PR (7) | 79.5 |
| 9 | Small lymphocytic (IV A) | 484 | 45 | 4 | 3 | N | CR (14) | CR (50+) | 106.5 |
| 10 | Transformed follicular (II B) | 308 | 32 | 4 | 1 | Y | PR (2) | PR (25) | 105.2 |
| 11 | Follicular (IV A) | 842 | 42 | > 5 | 2 | Y (refractory) | MR (6) | CR (45+) | 106.0 |
| 12 | Follicular (IV B) | 1451 | 73 | > 5 | 3 | Y | PR (11) | PR (25) | 95.6 |
| 13 | Follicular (IV A) | 127 | 37 | 3 | 1 | Y (refractory) | PR (3) | CR (41+) | 141.3 |
| 14 | Follicular (IV A) | 676 | 53 | > 5 | 3 | N | PR (7) | CR (28+) | 131.8 |
| 15 | Follicular (III A) | 169 | 23 | > 5 | 2 | N | PR (3) | CR (27+) | 138.72 |
| 16 | Follicular (IV B) | 679 | 58 | > 5 | 2 | N | CR (22) | PR (20) | 128.3 |
Evaluation of disease response
Disease status was evaluated by physical examination, serial CT scanning, and bone marrow biopsies (if bone marrow was involved at baseline) at 3, 6, and 12 months after RIT, and then as clinically indicated. Follow-up was continued for 24 months after the start of the study protocol for all patients. Response evaluation was in accordance with the International Workshop of Standardized Response Criteria for NHL.24
Evaluation of treatment toxicity
Adverse events were classified according to the Common Terminology Criteria for Adverse Events, version 3.0.25 Full blood counts were carried out weekly from initiation of treatment until 13 weeks after the last therapeutic dose, then every 3 months for 2 years. Hepatic and renal function was assessed weekly for 6 weeks, then every 3 months. Thyroid function was monitored at 3-month intervals.
Results
Induction therapy with rituximab increases the effective half-life of subsequent 131I-rituximab RIT
The schema for the clinical trial is shown in Figure 1A with an induction dose of 4 weekly infusions of 375 mg/m2 rituximab followed by a further 100 mg/m2 rituximab before the dosimetric and therapeutic 131I-rituximab. The effective half-life of 131I-rituximab was calculated from the decline in whole-body activity recorded during 3 sequential gamma camera scans. Figure 1B shows the pharmacokinetics of a 185-MBq dosimetric tracer of 131I-rituximab administered to 5 patients (taken from cohorts 2 and 3). They clearly demonstrate a much shorter effective half-life before receiving any rituximab. Treatment with 4 weekly doses of rituximab followed by a further 100 mg/m2 predose significantly increases the effective half-life of 131I-rituximab. The mean effective half-life of 131I-rituximab increased from 45.04 hours (range, 40.8-55.2 hours) in the absence of any prior rituximab, to 92.56 hours (range, 66.5-113.6 hours) in the same 5 patients after induction with 4 weekly infusions of unlabeled rituximab (P = .003). The effective half-lives of the first and second dosimetric tracers of 131I-rituximab at week 7 and week 15 were calculated for all 16 patients after 4 weekly infusions. For the whole group, similar pharmacokinetics are seen between the first and the second infusions after treatment with rituximab (Figure 1B). The effect of the induction treatment with rituximab on the biodistribution of 131I-rituximab is illustrated by 2 representative gamma camera scans.In the absence of any prior rituximab, the radioimmunoconjugate localizes to the spleen(Figure 1C); whereas after treatment with rituximab, the biodistribution is improved with targeting to tumor and more widespread distribution throughout the body (Figure 1D).

131I-rituximab administration. (A) Schematic outline of treatment schedule for rituximab followed by 131I-rituximab. A total of 375 mg/m2 of unlabeled rituximab was administered weekly for 4 weeks, shown as weeks 0, 1, 2, and 3. The dosimetric tracers of 185 MBq of 131I-rituximab were delivered on weeks 7 and 15. The therapeutic fractions of 131I-rituximab were each administered 2 weeks after dosimetry, at weeks 9 and 17. Both the dosimetric tracers and the therapeutic fractions of 131I-rituximab were delivered immediately after an infusion of 100 mg/m2. The dosimetry of 5 patients was assessed with a 185-MBq dosimetric tracer dose of 131I-rituximab before receiving any rituximab. (B) The clearance of 131I-rituximab in patients before rituximab and after the first and second 131I-rituximab tracer dose. The pharmacokinetics of 131I rituximab were calculated using whole-body anterior and posterior gamma camera images obtained within 1 hour after completion of the tracer infusion dose and on days 4 and 6 after 131I-rituximab administration. For the prerituximab group, no rituximab had been administered before the dosimetric studies. For groups marked “dose 1” and “dose 2,” 4 weekly infusions of 375 mg/m2 were given according to the schedule in panel A. For the predose, a further 100 mg/m2 of unlabeled rituximab was administered as an intravenous infusion, followed 2 to 3 hours later by an infusion of 185 MBq 131I-rituximab. (C) 131I-rituximab administered without prior rituximab treatment is mostly sequestered to the spleen. Whole-body gamma camera scanning was performed after administration of 185 MBq 131I-rituximab without any prior rituximab. The radiolabeled rituximab and gamma activity is visualized within the spleen. (D) Biodistribution of 131I rituximab after 4 loading doses of rituximab. After induction therapy with 4 weekly infusions of rituximab (375 mg/m2), the 131I-rituximab no longer localizes to the spleen, and there is a more widespread biodistribution throughout the body. Tumor targeting within known pelvic lymph nodes is observed.
131I-rituximab administration. (A) Schematic outline of treatment schedule for rituximab followed by 131I-rituximab. A total of 375 mg/m2 of unlabeled rituximab was administered weekly for 4 weeks, shown as weeks 0, 1, 2, and 3. The dosimetric tracers of 185 MBq of 131I-rituximab were delivered on weeks 7 and 15. The therapeutic fractions of 131I-rituximab were each administered 2 weeks after dosimetry, at weeks 9 and 17. Both the dosimetric tracers and the therapeutic fractions of 131I-rituximab were delivered immediately after an infusion of 100 mg/m2. The dosimetry of 5 patients was assessed with a 185-MBq dosimetric tracer dose of 131I-rituximab before receiving any rituximab. (B) The clearance of 131I-rituximab in patients before rituximab and after the first and second 131I-rituximab tracer dose. The pharmacokinetics of 131I rituximab were calculated using whole-body anterior and posterior gamma camera images obtained within 1 hour after completion of the tracer infusion dose and on days 4 and 6 after 131I-rituximab administration. For the prerituximab group, no rituximab had been administered before the dosimetric studies. For groups marked “dose 1” and “dose 2,” 4 weekly infusions of 375 mg/m2 were given according to the schedule in panel A. For the predose, a further 100 mg/m2 of unlabeled rituximab was administered as an intravenous infusion, followed 2 to 3 hours later by an infusion of 185 MBq 131I-rituximab. (C) 131I-rituximab administered without prior rituximab treatment is mostly sequestered to the spleen. Whole-body gamma camera scanning was performed after administration of 185 MBq 131I-rituximab without any prior rituximab. The radiolabeled rituximab and gamma activity is visualized within the spleen. (D) Biodistribution of 131I rituximab after 4 loading doses of rituximab. After induction therapy with 4 weekly infusions of rituximab (375 mg/m2), the 131I-rituximab no longer localizes to the spleen, and there is a more widespread biodistribution throughout the body. Tumor targeting within known pelvic lymph nodes is observed.
The effective half-life increases markedly between delivery of the first and second fractions of 131I-rituximab in patients with larger volumes of disease
Figure 2A demonstrates the increase in effective half-life of 131I-rituximab after induction doses of rituximab. Although for the group of patients as a whole there appeared to be no change in the effective half-life between the first and the second dosimetric study, in some patients there were marked increases in the effective half-life between the first and the second infusion. We next analyzed these data according to the tumor burden of the patients at the time of treatment. Before therapy splenic and lymph node volumes were calculated radiologically for each patient from whole-body cross-sectional CT scanning, as described in “Methods.” The tumor volumes ranged from 147 to 1494 mL and are shown for each patient in Table 2. Despite the apparent similarity of the effective half-life for all patients between the first and second dosimetric infusions, there were, however, large differences between the first and second infusion in those patients who presented with large volumes of tumor. In 4 patients, the effective half-life of the 131I-rituximab in the second biodistribution studies increased by more than 15% above the half-life observed with the first tracer infusion. Patients with the largest splenic and lymph node volumes showed the largest increases in effective half-life (P = .007; Figure 2B). Those patients with the highest initial tumor volumes, for example, patients 4 and 12 (1494 mL and 1451 mL, respectively), increased the effective half-lives of the second tracer of 131I-rituximab by 43.3% and 40%, respectively.

Tumor burden. (A) Sequential dosimetric studies illustrating the increase in effective half-life of 131I rituximab after prior rituximab and between the first and second dosimetric doses. All 5 patients who had not received prior rituximab demonstrate an increase in the mean effective half-life of 131I rituximab from 45 to 98 hours. After rituximab treatment, the majority of patients show no change in the effective half-life between the first and the second dosimetric study; however, 4 patients demonstrate a marked increase in the effective half-life between the first and the second infusion. (B) The effect of tumor burden on the effective half-life of 131I-rituximab. Patients with greater tumor burden demonstrate significant increases in effective half-life of 131I-rituximab between delivery of the first and the second fractions. Each data point represents values for an individual patient. As the splenic and lymph node volume increases, the relationship between the effective half-life of the first and second dosimetric doses of 131I-rituximab significantly alters. With volumes of tumor of less than 600 mL, the effective half-lives for the first and second tracer infusions of 131I-rituximab are similar. In contrast, for patients with larger volumes of tumor and those with volumes more than 600 mL, there appears to be a substantial increase in the effective half-life of 131I-rituximab between delivery of the first and second fractions (P = .007).
Tumor burden. (A) Sequential dosimetric studies illustrating the increase in effective half-life of 131I rituximab after prior rituximab and between the first and second dosimetric doses. All 5 patients who had not received prior rituximab demonstrate an increase in the mean effective half-life of 131I rituximab from 45 to 98 hours. After rituximab treatment, the majority of patients show no change in the effective half-life between the first and the second dosimetric study; however, 4 patients demonstrate a marked increase in the effective half-life between the first and the second infusion. (B) The effect of tumor burden on the effective half-life of 131I-rituximab. Patients with greater tumor burden demonstrate significant increases in effective half-life of 131I-rituximab between delivery of the first and the second fractions. Each data point represents values for an individual patient. As the splenic and lymph node volume increases, the relationship between the effective half-life of the first and second dosimetric doses of 131I-rituximab significantly alters. With volumes of tumor of less than 600 mL, the effective half-lives for the first and second tracer infusions of 131I-rituximab are similar. In contrast, for patients with larger volumes of tumor and those with volumes more than 600 mL, there appears to be a substantial increase in the effective half-life of 131I-rituximab between delivery of the first and second fractions (P = .007).
Large splenic and lymph node volumes correlate with low serum rituximab levels and decreased effective half-life of 131I-rituximab
Serum rituximab levels were measured by ELISA before and after each rituximab infusion, corresponding to trough and peak levels for each patient. We observed that patients with low splenic and lymph node volumes demonstrated sequential increases in trough levels of serum rituximab, whereas patients with higher volumes of disease virtually cleared rituximab from the serum compartment after each dose. The effect of splenic and lymph node volume on serum rituximab levels is well illustrated in Figure 3A, which shows a comparison of the serum rituximab levels of representative patients with both low tumor volume (patient 2; volume = 147 mL) and larger tumor volumes (patient 4; volume = 1494 mL) over the course of the treatment phase and beyond. Trough levels of serum rituximab and splenic and lymph node volume were significantly negatively correlated (P = .006 at week 1, P = .003 at week 2, P = .019 at week 3), and this remained significant at week 7, when the first tracer dose of 131I-rituximab was administered (P = .016). Patients with low trough levels of serum rituximab demonstrated shorter effective half-lives of the 131I-rituximab tracer in the biodistribution study performed at week 7 (Spearman rank correlation coefficient = 0.700, P = .009). Interestingly, at serum rituximab levels less than 50 μg/mL, the effective half-life of 131I-rituximab appears to decline rapidly, whereas for patients whose serum level is more than 50 μg/mL, it appears to reach a plateau of approximately 100 hours (Figure 3B).
![Figure 3. Serum rituximab relationships. (A) The relationship between serum rituximab levels and tumor burden. The difference in serum rituximab levels between patients with low and high tumor volumes is illustrated. Serum rituximab concentrations were measured throughout the treatment protocol and are plotted for patient 2 (calculated spleen and lymph node volume [CSLV] = 147 mL) and patient 4 (CSLV = 1494 mL). Both patients were administered loading doses of 375 mg/m2 rituximab at weeks 0, 1, 2, and 3. Patients with high tumor volumes, as represented by patient 4, saw near-complete clearance of serum rituximab after each loading dose, and their serum rituximab level remained low when the first dosimetric tracer of 131I-rituximab was administered at week 7. For those with low tumor volumes, however, there was a sequential increase in the peak and trough levels of rituximab throughout the loading process, and serum rituximab concentrations remained significantly elevated in these patients at week 7. ♦ represents patient 2; ■, patient 4; +, administration of unlabeled rituximab (375 mg/m2), * Administration of 131I-rituximab and unlabeled rituximab (100 mg/m2). (B) The relationship between serum rituximab level and effective half-life of 131I-rituximab. Serum levels of rituximab were measured at week 7, and low levels correlate with a shorter effective half-life of the first dosimetric tracer of 131I-rituximab (Spearman rank correlation coefficient = 0.700, P = .009). For patients with serum rituximab levels of less than or equal to 50 μg/mL, the effective half-life of 131I-rituximab appears to rapidly decline; whereas for patients whose serum level is more than 50 μg/mL, the half-life of the radioimmunoconjuate appears to reach a plateau of approximately 100 to 120 hours.](/view-large/figure/7014412/zh80100931810003.jpeg)
Serum rituximab relationships. (A) The relationship between serum rituximab levels and tumor burden. The difference in serum rituximab levels between patients with low and high tumor volumes is illustrated. Serum rituximab concentrations were measured throughout the treatment protocol and are plotted for patient 2 (calculated spleen and lymph node volume [CSLV] = 147 mL) and patient 4 (CSLV = 1494 mL). Both patients were administered loading doses of 375 mg/m2 rituximab at weeks 0, 1, 2, and 3. Patients with high tumor volumes, as represented by patient 4, saw near-complete clearance of serum rituximab after each loading dose, and their serum rituximab level remained low when the first dosimetric tracer of 131I-rituximab was administered at week 7. For those with low tumor volumes, however, there was a sequential increase in the peak and trough levels of rituximab throughout the loading process, and serum rituximab concentrations remained significantly elevated in these patients at week 7. ♦ represents patient 2; ■, patient 4; +, administration of unlabeled rituximab (375 mg/m2), * Administration of 131I-rituximab and unlabeled rituximab (100 mg/m2). (B) The relationship between serum rituximab level and effective half-life of 131I-rituximab. Serum levels of rituximab were measured at week 7, and low levels correlate with a shorter effective half-life of the first dosimetric tracer of 131I-rituximab (Spearman rank correlation coefficient = 0.700, P = .009). For patients with serum rituximab levels of less than or equal to 50 μg/mL, the effective half-life of 131I-rituximab appears to rapidly decline; whereas for patients whose serum level is more than 50 μg/mL, the half-life of the radioimmunoconjuate appears to reach a plateau of approximately 100 to 120 hours.
Serum rituximab relationships. (A) The relationship between serum rituximab levels and tumor burden. The difference in serum rituximab levels between patients with low and high tumor volumes is illustrated. Serum rituximab concentrations were measured throughout the treatment protocol and are plotted for patient 2 (calculated spleen and lymph node volume [CSLV] = 147 mL) and patient 4 (CSLV = 1494 mL). Both patients were administered loading doses of 375 mg/m2 rituximab at weeks 0, 1, 2, and 3. Patients with high tumor volumes, as represented by patient 4, saw near-complete clearance of serum rituximab after each loading dose, and their serum rituximab level remained low when the first dosimetric tracer of 131I-rituximab was administered at week 7. For those with low tumor volumes, however, there was a sequential increase in the peak and trough levels of rituximab throughout the loading process, and serum rituximab concentrations remained significantly elevated in these patients at week 7. ♦ represents patient 2; ■, patient 4; +, administration of unlabeled rituximab (375 mg/m2), * Administration of 131I-rituximab and unlabeled rituximab (100 mg/m2). (B) The relationship between serum rituximab level and effective half-life of 131I-rituximab. Serum levels of rituximab were measured at week 7, and low levels correlate with a shorter effective half-life of the first dosimetric tracer of 131I-rituximab (Spearman rank correlation coefficient = 0.700, P = .009). For patients with serum rituximab levels of less than or equal to 50 μg/mL, the effective half-life of 131I-rituximab appears to rapidly decline; whereas for patients whose serum level is more than 50 μg/mL, the half-life of the radioimmunoconjuate appears to reach a plateau of approximately 100 to 120 hours.
Clinical efficacy of fractionated 131I-rituximab
Table 2 summarizes the clinical response to treatment for all patients. The ORR was 94% with 50% (8 of 16) achieving CR and 44% (7 of 16) PR; one patient showed a minor response. The response rate is impressive in a group of heavily pretreated patients with intermediate to poor Follicular Lymphoma International Prognostic Index (FLIPI) scores. Time to progression for the treatment group is presented as a Kaplan-Meier plot (Figure 4), and the median time to progression from the start of the study protocol was 20 months. In contrast, median progression-free survival for the patient group after their previous chemotherapy regimens was 6.5 months. Time to progression was significantly longer with fractionated 131I-rituximab than with the last qualifying chemotherapy (P < .001).

Progression-free survival curves. Curve A indicates after last qualifying chemotherapy; curve B, after fractionated 131I-rituximab.
Progression-free survival curves. Curve A indicates after last qualifying chemotherapy; curve B, after fractionated 131I-rituximab.
Safety and tolerability of fractionated 131I-rituximab
Fractionated RIT with 131I-rituximab was well tolerated by all patients throughout the study, and no dose-limiting toxicities were observed. No serious adverse events or infusional reactions were reported during the treatment schedule. Two patients in the 2 lower dose cohorts A and B developed reversible grade 4 neutropenia during the treatment phase, with nadir neutrophil counts of 0.2 × 109 cells/L and 0.3 × 109 cells/L, respectively, although this occurred earlier than would have been expected if secondary to RIT, and no episodes of neutropenia were seen with the higher radiation doses. There were no episodes of neutropenic sepsis, and it is notable that there was no grade 3/4 anemia, neutropenia, or thrombocytopenia observed in the 2 highest radiation dose cohorts. Figure 5 illustrates the lack of myelotoxicity in the highest dose cohort D, all of whom received more than 125 cGy cumulative WBD. No patients in the study required blood product or growth factor support during the study period.

Hemoglobin concentrations, neutrophil counts, and platelet counts. (A) Hemoglobin concentrations (g/L). (B) Neutrophil counts (109/L). (C) Platelet counts (109/L) for cohort D over a 40-week time period. Cohort D was the maximal dose cohort in this protocol, with each patient receiving a maximal cumulative whole-body dose greater than 125 cGy in 2 fractions of 131I-rituximab.
Hemoglobin concentrations, neutrophil counts, and platelet counts. (A) Hemoglobin concentrations (g/L). (B) Neutrophil counts (109/L). (C) Platelet counts (109/L) for cohort D over a 40-week time period. Cohort D was the maximal dose cohort in this protocol, with each patient receiving a maximal cumulative whole-body dose greater than 125 cGy in 2 fractions of 131I-rituximab.
Discussion
This study is the first to undertake a detailed pharmacokinetic analysis of radiolabeled anti-CD20 mAb and to simultaneously examine both the influence of tumor burden and repeated dosing with unlabeled or “cold” rituximab on the efficacy and toxicity of RIT. Several important findings have emerged from this work. First, the effective half-life of 131I-rituximab doubled from approximately 45 hours to more than 90 hours after induction therapy with rituximab. Second, low serum levels of rituximab after induction and predosing with rituximab correlated with a shorter effective half-life of 131I-rituximab. Third, the half-life of the radioimmunoconjugate increased between the first and second fraction of RIT in patients with large volumes of tumor. The large interpatient and intrapatient variation suggests that dosimetry is important for optimal delivery of 131I anti-CD20 RIT. Fourth, we found that induction therapy with multiple doses of rituximab did not appear to compromise the clinical efficacy of subsequent radioimmunotherapy with 131I-rituximab, with high response rates leading to durable remissions that were greatly superior to the previous chemotherapy response durations. Finally, 2 fractionated doses of 131I-rituximab allowed high cumulative doses to be delivered without significant hematologic toxicity, and the toxicity of radiolabeled chimeric mAb was no greater than that seen with murine radiolabeled mAb.
The pharmacokinetics of rituximab have previously been studied and found to be influenced by the availability of CD20 (the “antigen sink”), with the greater number of CD20 binding sites in bulky disease causing sequestration of rituximab and decreasing concentrations in the serum compartment.26 We observed a significant negative correlation between splenic and lymph node volumes and trough serum levels of rituximab after initial rituximab dosing. This result is in agreement with previous pharmacokinetic studies of rituximab, which found that serum concentrations were lower in patients with high tumor burdens.27,28 The present study takes these observations further by determining the effective half-life of 131I-rituximab using sequential gamma camera images. Unlike ELISA measurements of serum rituximab, the dosimetry calculations detect whole-body 131I activity, regardless of whether the radioimmunoconjugate is bound to CD20 or circulating. Therefore, decreased effective half-life in circumstances of increased CD20 availability cannot be explained by sequestration of 131I-rituximab away from the serum compartment. The decreased effective half-life of the radioimmunoconjugate has to instead be secondary to accelerated elimination of 131I and gamma activity from the body. A potential explanation for our findings whereby clearance of 131I is accelerated in circumstances of increased CD20 availability is that binding of 131I-rituximab to CD20 promotes the degradation of 131I-rituximab, with deiodination and renal excretion of free 131I. The CD20 antigen has previously not been thought to modulate on binding anti-CD20 antibodies using in vitro assays.29,30 However, it has been postulated that rituximab in vivo may be phagocytosed, degraded, or obscured as part of an antibody-dependent cellular cytotoxicity response, or deposition of C3 fragments during complement activation, both of which may be triggered by rituximab binding.31-34
An interesting observation to emerge from this study was that patients with large initial tumor burdens at study entry exhibited a greater than 40% increase in 131I-rituximab's effective half-life between delivery of the first and the second fractions. We suggest that the most likely explanation for these large differences in the effective half-life between infusions is secondary to the dramatic tumor responses that were observed clinically in these patients after the first therapeutic fraction of 131I-rituximab. Such a reduction in tumor load would thus reduce the CD20 tumor “antigen sink” in bulky disease. Significant interpatient variability has been previously demonstrated in the pharmacokinetics of 131I-rituximab,22,35,36 and others have administered the dose of 131I-rituximab according to the results of prospective dosimetry.37 This study, however, provides new insights into how the effective half-life can substantially alter over the course of a few weeks between the first and the second fraction of RIT. Here we demonstrate that, with 2 fractions of RIT, the effective half-life of 131I-rituximab is subject not only to significant interpatient variability, but that large differences in dosimetry can be seen between 2 fractions in the same patient, and this is especially the case for those patients with high tumor burdens. Therefore, dosimetry before administration of each fraction of 131I-rituximab appears to be essential before each infusion for accurate WBD titration.
Predosing with unlabeled or “cold” anti-CD20 mAb has become standard practice in RIT targeting the CD20 antigen.22,38 The predose is thought to increase tumor targeting of the labeled mAb by blocking “nonspecific” binding sites, such as circulating and splenic B cells. The initial gamma camera scans in this study demonstrated that, without predosing, the administered radioimmunoconjugate was mostly sequestered in the spleen. After 4 weekly infusions of 375 mg/m2 rituximab, a more favorable biodistribution was seen (Figure 1C,D) with selective tumor targeting in many patients at sites of nodal disease (data not shown).
Recent clinical data from the FIT study have provided compelling evidence for the efficacy of RIT after induction chemotherapy.10 However, the majority of patients in the FIT study did not receive rituximab-containing regimens as part of their initial treatment. Therefore, an important question in current clinical practice is whether repeated doses of rituximab will prevent subsequent binding of radiolabeled anti-CD20 antibody to tumor sites, potentially decreasing efficacy and causing toxicity by increasing normal tissue radiation dose. Furthermore, the optimal predosing regimen with “cold” antibody before RIT has not been fully determined and the widespread use of rituximab in induction therapy as part of immuno-chemotherapy provides a further potential confounding factor if RIT is used as a consolidation therapy after rituximab-containing regimens. The early studies establishing the utility of a predose of unlabeled or “cold” mAb were based on relatively small patient numbers.7,39 For 90Y-ibritumomab (Zevalin), the current predosing regimen was based on just 6 patients. In this study, no differences were observed in biodistribution between the 3 patients who received 125 mg/m2 and the next 3 patients who were given 250 mg/m2. On the basis of these results, 250 mg/m2 became the predose used for imaging and therapeutic infusions. The amount of radiolabeled mAb protein administered is considerably smaller than the predoses of unlabeled mAb for both 90Y-ibritumomab tiuxetan and 131I-tositumomab regimens. Just 3.2 mg of 90Y-ibritumomab is used after a 250 mg/m2 predose of rituximab, and 35 mg of 131I-tositumomab after a 450 mg unlabeled predose of tositumomab.38-41
Recently, concerns have been expressed that repeated doses of rituximab would lead to high serum levels of the mAb, which in turn would saturate the CD20 antigens in patients with small tumor volumes. Saturation of CD20 antigen by rituximab might in turn then result in unbound radioimmunoconjugate having a longer effective half-life and slower clearance from the body, resulting in increased normal tissue toxicity. Preclinical evidence supporting the concept that rituximab might block the binding of the radioimmunoconjugate has recently been published in a xenograft model.11,42 The caveats in extrapolating such a model to the clinical situation include the lack of cross-reactivity of rituximab with normal host B cells. In contrast to this preclinical data, however, recent phase 2 clinical data using several doses of induction therapy with rituximab alone or as part of R-CHOP appear to confirm that subsequent anti-CD20 RIT binds to tumor with good clinical efficacy.43 Our study is the first to address this specific question in a clinical situation, and our results suggest that administration of induction therapy with rituximab does not compromise the antitumor efficacy of radiolabeled anti-CD20 or increase the myelotoxicity. We observed no relationship between the pharmacokinetics of rituximab in individual patients and the clinical responses elicited by fractionated 131I-rituximab. The median time to progression after fractionated 131I-rituximab was 20 months in a group of heavily pretreated patients (Table 1). The time to disease progression in these patients after fractionated 131I-rituximab was significantly longer than that seen after their last qualifying chemotherapy (P < .001). Table 2 shows the response and response durations to the last line of chemotherapy compared with that seen with the fractionated RIT. Three patients with transformed follicular lymphoma were included in the study, one of whom was refractory to chemotherapy (patient 8) and a second who had progressed just 2 months after completing chemotherapy (patient 10). After the fractionated RIT, the drug refractory case (patient 8) responded with a partial response of 7 months' duration. Patient 10 had a 25-month remission after the fractionated RIT regimen. Seven patients previously treated with rituximab were included in the study, and 4 of these patients relapsed within 6 months of receiving the drug, fulfilling the US FDA definition of rituximab refractory. It is interesting to note that 3 of these 4 rituximab-refractory patients continue to enjoy ongoing CR, from between 41 to 60 months after completing fractionated 131I rituximab.
This fractionated 131I-rituximab regimen had an excellent safety profile with surprisingly little hematologic toxicity, given the high cumulative whole-body radiation doses achieved in the highest dose cohorts. No patient developed grade 3 or 4 thrombocytopenia, and only 2 patients developed transient grade 4 neutropenia. However, in these 2 isolated cases of neutropenia, it is perhaps important to note that the observed nadir did not correspond with the kinetics of the expected delayed nadir after RIT with 90Y-ibritumomab.39 Furthermore, the neutropenia did not correspond with thrombocytopenia, as would be expected if the effect was secondary to targeted radiation administered with the therapeutic RIT. Therefore, it seems doubtful that the isolated neutropenia seen in the lower radiation dose cohorts was directly related to the targeted radiation but was instead secondary to the unlabeled rituximab. Rituximab-induced neutropenia is an increasingly recognized, but poorly understood, reaction to the drug.44 The 4 patients in cohort D received a mean WBD of 135 cGy over the treatment schedule, and none exhibited dose-limiting hematologic or nonhematologic toxicity (Figure 3). The toxicity profile of fractionated 131I-rituximab in this study compares favorably with 90Y-ibritumomab tiuxetan and 131I-tositumomab RIT, where reversible grade 4 neutropenia was seen in 32% and 16% of treated individuals, respectively.45,46 Both 90Y-labeled ibritumomab tiuxetan (Zevalin) and 131I-labeled tositumomab (Bexxar) radioimmunoconjugates are murine mAbs and were preferred in the development of RIT because of the shorter half-life of the murine mAb. A theoretical concern with using chimeric mAb is that the increased half-life of the chimeric mAb would increase the overall radiation dose to bone marrow and result in increased myelotoxicity.47 This study, however, demonstrates that the longer half-life of the chimeric antibody can be safely used in RIT and does not lead to increased myelotoxicity despite whole-body doses given in 2 fractions of more than 120 cGy.
In conclusion, this is the first study to relate the pharmacokinetics of serum rituximab to the burden of lymphoma as well as correlating these data to the subsequent dosimetry of anti-CD20 RIT. We have found that repeated rituximab dosing increases the effective half-life of 131I-rituximab and alters its biodistribution, but importantly this does not appear to compromise the clinical efficacy or increase the normal tissue toxicity. Fractionated RIT using 131I-rituximab is well tolerated and allows the delivery of much higher WBD in 2 fractions than has previously been achieved with single fraction RIT. The effective half-life of 131I-rituximab is influenced by the tumor volume and the serum rituximab level, with substantial interpatient and intrapatient variation over the course of the treatment protocol, emphasizing the importance dosimetry has in patient specific delivery of RIT. The clinical responses in this study compare very favorably with previous studies of single-dose RIT and with multiple cycles of immunochemotherapy now used as “standard” therapy in relapsed indolent NHL.48 These promising results form the basis for further studies using fractionated RIT, which we have initiated and are ongoing within Europe. Finally, we have confirmed that the clinical efficacy of anti-CD20 RIT does not appear to be compromised by previous repeated dosing with rituximab and thus provide evidence for the clinical community that RIT can be used effectively after multiple doses of rituximab.
Preliminary data were presented orally at the American Society of Hematology Annual Meeting, San Diego, CA, December 2004, and at the 9th International Conference on Malignant Lymphoma, Lugano, Switzerland, June 2005.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the members of the Southampton Cancer Research United Kingdom Clinical Trial Center for their help with coordination and organization of the trial, Dr Ric Swindell for assistance with statistical analysis, and members of the Radiation Related Research Group, Manchester Cancer Research Center for helpful advice.
This work was supported by Cancer Research UK (grants C431/A3313 and C328/A2738), Wessex Cancer Trust, and the United Kingdom Department of Health through the Experimental Cancer Medicine Network.
Authorship
Contribution: T.M.I., M.B., S.C., M.S.C., M.J.G., J.S., and P.W.M.J. designed and conducted the trial; T.M.I., M.B., and N.S.B. performed data analysis; T.M.I. and N.S.B. wrote the manuscript; M.Z. performed whole-body dose calculations; and J.T., Y.D., V.L., and M.Z. prepared the 131I-rituximab and calculated effective half-lives.
Conflict-of-interest disclosure: T.M.I. has received honorarium as part of speakers' bureau and for consultancy work with Bayer Schering Pharma. P.W.M.J. has received honorarium as part of speakers' bureau for Roche Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Tim M. Illidge, CRUK Targeted Therapy Group, School of Cancer and Imaging Sciences, Manchester Cancer Research Centre, University of Manchester, Wilmslow Road, Manchester, United Kingdom, M20 4BX; e-mail: tmi@manchester.ac.uk.
