

Protein C (PC) deficiency increases the risk of venous thrombosis (VT) among members of Kindred Vermont II but fails to fully account for the inheritance pattern. A genome scan of the pedigree supported the presence of a prothrombotic gene on chromosome 11q23 (nominal P < .0001), with weaker support on chromosomes 10p12 (P < .0003) and 18p11.2-q11 (P < .0007). Resequencing of 109 genes in the linkage regions identified 5030 variants in a sample of 20 kindred members. Of 16 single nucleotide polymorphisms in 6 genes tested in the larger family set, only single nucleotide polymorphisms in cell adhesion molecule 1 (CADM1) associated with VT. Among the 8 CADM1 single nucleotide polymorphisms genotyped in the complete sample, rs6589488 was most strongly supported (P < .000007), but the association was limited to the PC-deficient subset of the sample (P < .000001). Haplotype analysis narrowed the region containing the causative variant to the coding region of the CADM1 gene. CADM1 gene expression analyzed in blood outgrowth endothelial cells cultured from family members was decreased compared with control subjects, lending phenotypic support to this conclusion. Finally, we have for the first time demonstrated CADM1 in endothelial cells, where it appears to be selectively involved in endothelial cell migration, suggesting a role in endothelial barrier repair.
Introduction
Inherited thrombophilia is characterized by the onset of recurrent venous thrombosis (VT) before the age of 50. The risk of VT in families with thrombophilia increases substantially with the cooccurrence of 2 or more risk factors.1,–3 Recently the researchers in the European Prospective Cohort on Thrombophilia (EPCOT) study4 estimated an adjusted relative risk of VT of 16.4 (confidence interval, 9.6-28.0) associated with familial thrombophilia, with the greatest incidence of VT observed in those with multiple genetic risk factors.
The known genetic risk factors for inherited thrombophilia include both variants in procoagulant proteins and deficiencies of coagulation inhibitors.5 The procoagulant protein variants factor V Leiden6 and prothrombin G20210A7 are the most common prothrombotic genetic risk factors, with a prevalence in white subjects of approximately 5% and 2%, respectively.8 The coagulation inhibitors with deficiencies that associate with thrombophilia include antithrombin, protein S, and protein C (PC).9,–11 Many different mutations produce PC deficiency, with a combined prevalence of 0.3%.12 Although at increased risk of VT, especial-ly in penetrant families,13 most PC-deficient persons are asymptomatic.14
PC deficiency in Kindred Vermont II results from a 3363 inserted (Ins) C mutation in exon 6 of the PC gene.15 Although 3363InsC increases risk of VT, it does not fully account for the pattern of inheritance.16,17 Further evidence that additional thrombophilia genes segregate in the kindred comes from an autosomal genome-wide linkage analysis that implicated chromosome 11q23 as well as potentially chromosomes 18p11.2-q11.2 and 10p12.18 We previously tested and rejected PAFAH1B2 as the chromosome 11q23 thrombophilia gene.19
Herein, we evaluate variants identified through resequencing of 109 genes within the linkage regions on chromosomes 10, 11, and 18 as candidates for thrombophilia genes that interact with PC deficiency in Kindred Vermont II. We have identified cell adhesion molecule 1 (CADM1), a member of the immunoglobulin superfamily 4, as a strong candidate gene for risk of thrombosis and show for the first time that it is present in endothelium. In addition we provide preliminary evidence supporting a role for CADM1 in maintaining endothelial barrier function.
Methods
Subjects
Kindred Vermont II includes all available descendants, plus spouses, of a couple born in the 1830s. The pedigree spans 7 generations, with data available on the most recent 5. Pedigree members, descendants of French Canadians and Abenaki American Indians, reside primarily in Vermont. Each diagnosis of VT was confirmed through objective tests performed during the subject's hospitalization for treatment of deep-vein thrombosis. The initial pedigree structure and data collection have been described more extensively elsewhere16 ; additional data collection in 2002 updated diagnoses and sampled later generations and descendants of a sister of the wife of the original couple. Expanded Kindred Vermont II includes an additional 8 pedigrees from Quebec, Canada and New Hampshire who share a common ancestor with the original kindred and have members who carry 3363InsC.20 These pedigrees span 2 to 4 generations, with 2 to 26 members studied. This study was approved by the Human Experimentation Committees of the University of Vermont College of Medicine, the Beth Israel Hospital, and Center Hospitalier Affilié Université Laval, and informed consent was obtained in accordance with the Declaration of Helsinki.
Resequencing
A total of 109 genes from the linkage regions on chromosomes 11q23, 18p11.2-q11.2, and 10p1218 were selected as potential thrombophilia candidates or to fill gaps between candidates. We classified as potential candidates genes involved in hemostasis, inflammation, or the regulation of either pathway. Resequencing of the 109 genes from 20 extended kindred members (15 affected, 5 unaffected) was performed at the J. Craig Venter Institute through the NHLBI Genotyping and Resequencing Service (http://rsng.nhlbi.nih.gov/scripts/index.cfm). The 15 affected persons to be resequenced were selected for a first thrombotic event at an early age and to be distantly related, thereby minimizing their sharing of other genomic regions; the 5 unaffected persons were selected to have thrombosis risk factors (older age, PC deficiency) and to be distantly related. Resequencing of each gene included 2 kb upstream of the 5′ most exon, full exon coverage (includes intron/exon boundaries), conserved noncoding regions determined by use of the UCSC Genome Bioinformatics database, and 2 kb downstream of the 3′ most exon. Thus, we have sequence for coding regions, noncoding regulatory regions, and splice junction regions for each of 109 candidate genes.
Single nucleotide polymorphism genotyping
The genotyping of single nucleotide polymorphisms (SNPs) to validate the resequencing results was performed in the genotyping core laboratory in the Laboratory for Clinical and Biochemical Research at the University of Vermont College of Medicine. SNPs were genotyped by the use of TaqMan Assays By Design (Applied Biosystems) under standard conditions (TaqMan SNP Genotyping Assays Protocol, Rev B, Part #4332856b, Applied Biosystems). End-point fluorescence values were scored manually by use of the SDS software (Applied Biosystems).21
Genetic analysis
Likelihood analysis, as implemented in jPAP (http://hasstedt.genetics.utah.edu/, Version 1.7.0),22 was used for all parameter estimation and hypothesis testing. Ascertainment was corrected by dividing the likelihood of each pedigree by the probability of a single affected, PC-deficient proband. Parameters were estimated as the values that maximized the likelihood. Hypotheses were tested through comparison of the maximized likelihoods of general and nested models. Asymptotically, and under certain regularity conditions, twice the natural logarithm of the likelihood ratio distributes as a χ2, or as a mixture of χ2 for a 1-tailed test when the nested model constrains a parameter at its boundary.23 The degree of freedom for each test equals the number of parameters constrained by the nested model.
Risk of thrombosis was modeled to account for age at first thrombotic event in affected pedigree members, while allowing for censored observations, through a modification of the age-of-onset regressive logistic model,24 also known as the age at diagnosis regressive model,25 and described as Method 2 in Cui et al.26 Let W represent age at first thrombotic event or age last examined if unaffected and X = 0/1 for male/female. The logit of the probability of thrombosis equals the following:
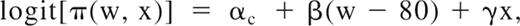
where π(w, x) = Pr(T = 1|W = w, X = x) denotes the probability of thrombosis and c denotes PC deficient/normal, pc = ln(αc/(1 − αc) represents the lifetime penetrance, exp(β) represents the annual odds ratio caused by age, and exp(γ) represents the female/male odds ratio. Parameter p varies from 0 to 1; parameters β and γ vary from 0 to ∞. Previous implementations of this model assumed the logistic density; the implementation in jPAP assumed the normal density to allow inclusion of a polygenic component parameterized as heritability specific for PC deficient/normal (hc2); the genetic correlation between PC-deficient and -normal persons was assumed to equal 1.
To test each SNP for association with thrombosis, we coded covariates for dominant (Y = 0/1 for nonrisk allele homozygotes/other genotypes) and recessive (Z = 1/0 for risk allele homozygotes/other genotypes) effects of the SNP and added δcY + λcZ to the basic model, where exp(δc) and exp(λc) represent the odds ratios of carrying the risk allele and of being homozygous for the risk allele, respectively, by PC deficient/normal. 2-df tests of significance compared the model with δd = δn and λd = λn to the nested model with δd = δn = λd = λn = 0; separate 2-df tests of significance within normal/deficient PC compared the general model to the nested model with δc = λc = 0.
To test whether rs6589488 is causative, we assumed 2 tightly linked loci where the first locus was rs6589488 with alleles A and T and the second locus represented the causative variant and was specified by the basic model extended to 2 alleles and genotype-specific penetrance; dominance was assumed in the PC-deficient subset (pd1, pd2 = pd3 for genotypes 1, 2, 3) and equal penetrance was assumed in the normal PC subset (pn1 = pn2 = pn3 for genotypes 1, 2, 3). The allele frequency of rs6589488 was fixed at its estimate; the variant frequency was conditioned on rs6589488 allele (fA, fT). fA = 1, fT = 0 corresponded to rs6589488 being causative or in complete linkage disequilibrium with the causative variant. Confidence intervals that excluded fA = 1 or fT = 0 will reject rs6589488 as causative.
Tissue procurement and cell lines
Greater saphenous veins were collected from patients undergoing coronary artery bypass grafting at Fletcher Allen Health Care. The veins were processed as previously described.27
Human umbilical vein endothelial cells (HUVECs, 001-F) were purchased from Allcells, Inc and grown according to manufacturer's instructions. The cells were grown in flasks and plated onto glass cover slips coated with gelatin (Allcells Inc) under sterile conditions at 37°C, 5% CO2 in HUVEC Basal Medium containing HUVEC Basal Medium Supplement (Allcells Inc). Cells were subcultured when they reached confluence.
Immunohistochemistry and immunocytochemistry
The tissue sections were subjected to routine processing for immunofluorescence, including deparaffinization, rehydration, and antigen retrieval with 10 mmol/L sodium citrate. Slides were then blocked in preimmune donkey serum for 1 hour at room temperature in preparation for immunofluorescence staining. The sections were then incubated overnight at 4°C with chicken monoclonal anti-CADM1 IgY antibody (anti-SynCAM/TSLC1, clone 3E1, MBL Inc) or rabbit polyclonal IgG anti-CADM1 antibody (anti-SynCAM, Abcam). After rinses with buffer, the sections were incubated for 1 hour at room temperature with donkey anti–chicken IgY conjugated to CY2 (Jackson ImmunoResearch Laboratories) or donkey anti–rabbit IgG conjugated to Alexa 488, respectively (Invitrogen). Nuclei were visualized with 4′,6-diamidino-2-phenylindole hydrochloride for both cells and tissue.
Cultured HUVECs were permeabilized for 15 minutes with 0.1% Triton in phosphate-buffered saline, followed by blocking in 10% normal donkey serum for 1 hour at room temperature. The cells were then incubated for 1 hour at room temperature with a chicken monoclonal anti-CADM1 antibody (same as used for tissue sections) and a mouse monoclonal anti-CD31 antibody (clone JC70A, DAKO Inc) cocktail. After rinses with buffer, the cells were incubated with a donkey anti–chicken IgY conjugated to CY2 (Jackson Immunoresearch) and a donkey anti–mouse conjugated to Alexa 555 (Invitrogen) for 1 hour at room temperature. Phalloidin directly conjugated to Alexa 647 (Invitrogen) was used to identify actin in HUVECs. In separate experiments HUVECs were stained with anti–von Willebrand factor rabbit polyclonal antibody (DAKO Inc) with donkey anti–rabbit Alexa 488 as the secondary antibody following the procedure described previously. CD31 and von Willebrand factor staining were used to confirm endothelial cell phenotype of HUVECs.
The following controls for immunofluorescence were used for all experiments: a secondary negative control in which the slides were left in blocking buffer during primary antibody incubation and a positive control showing similar expression of CADM1 that used both chicken monoclonal and rabbit polyclonal antibodies for CADM1. A preabsorption assay completed for cell culture included a cocktail of CADM1 peptide (Abnova) and chicken monoclonal anti-CADM1 antibody at a proportion of 1:2, respectively. Detection of CADM1 was fully inhibited by preabsorption, whereas nonpreabsorbed cells showed positive staining.
Immunostained slides were imaged with a Zeiss LSM 510 META confocal scanning laser microscope (Zeiss MicroImaging). A Plan-Neofluar 25×/0.8 NA multi-immersion objective lens and a Plan Apochromat 63×/1.4 NA oil-immersion lens were used for imaging. All images were acquired as 12-bit in multitrack mode, with appropriate laser excitation and emission filters as previously described.27
Endothelial cell culture and RNA expression array
Blood outgrowth endothelial cells (BOECs) were cultured from 3 Vermont Kindred II subjects who traveled to Minneapolis to have blood drawn because shipping logistics could not meet the requirement that cultures be established within 9 hours of venipuncture. Venous blood (100 mL) was collected into 3.2% citrate (Vacuette, Greiner Bio-1), after the first few milliliters had been discarded. Buffy coat mononuclear cells were layered onto rat collagen I and cultured in a cocktail of endothelial growth factors exactly as previously described.28 Cultures were continued until approximately 5 × 107 BOECs were available. Quality control measures included morphologic and immunophenotype verification of endothelial identity and performance of cytogenetics so presence of culture-acquired clonal defects could be excluded.
From each culture, RNA was isolated and prepared exactly as previously described,29 and more than 15 μg of fragmented RNA was available. Samples were applied to the Affymetrix U133A chip, and hybridization and raw expression analysis were performed by the University of Minnesota Biomedical Imaging and Processing Facility.
As previously described,29 raw microarray data were analyzed and tested for significance of single-gene differences by the use of both the Welch t test and statistical analysis of microarray (SAM), a test that uses false discovery rate. The Welch test assumes a normal distribution of data; the SAM test was specifically developed for microarray data and small sample numbers, where normality is not known and multiple comparisons are taken into account. The data from the 3 Vermont subjects were compared with our concurrently obtained database of BOEC microarray expression from 27 healthy subjects of mixed sex, age, and race. The latter data have already been publicly posted.29 Original microarray data for this study have been deposited in the National Center for Biotechnology Information's Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE17078 and GSE9877 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17078 and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE9877).
Results
Genetic analysis
The sample comprised 1 large pedigree (designated the original kindred) plus 8 small distantly related pedigrees with members who inherited the identical PC mutation (3363InsC) from a common ancestor (collectively designated the extended kindred).20 Table 1 presents characteristics of the sample. Of 131 PC-deficient persons, 125 carry 3363InsC, and 9 (including 3 carriers of 3363InsC) carry 1 of 2 other PC mutations. The analysis treated all mutations identically because PC activity showed similar reductions in all mutation carriers. Because different thromobophilia genes that interact with PC deficiency may have been inherited in the distantly related pedigrees, we analyzed the original kindred separately as well as part of the extended kindred; insufficient data precluded separate analysis of the set of 8 small pedigrees.
Characteristics of the sample by affection status (verified thrombotic event) in the original and extended kindred
| Characteristic | Original kindred* | Extended kindred† | ||
|---|---|---|---|---|
| Affected | Unaffected | Affected | Unaffected | |
| N | 30 | 351 | 40 | 412 |
| Male/female | 14/16 | 155/196 | 21/19 | 180/232 |
| Age‡, y (SD) | 39.23 (17.52) | 34.65 (18.82) | 35.70 (16.75) | 35.50 (18.51) |
| PC deficient/normal | 23/7 | 61/290 | 33/7 | 98/314 |
| Characteristic | Original kindred* | Extended kindred† | ||
|---|---|---|---|---|
| Affected | Unaffected | Affected | Unaffected | |
| N | 30 | 351 | 40 | 412 |
| Male/female | 14/16 | 155/196 | 21/19 | 180/232 |
| Age‡, y (SD) | 39.23 (17.52) | 34.65 (18.82) | 35.70 (16.75) | 35.50 (18.51) |
| PC deficient/normal | 23/7 | 61/290 | 33/7 | 98/314 |
Large interconnected pedigree.
Large interconnected pedigree plus 8 distantly related small pedigrees.
Age at first thrombotic event if affected; age at time of study entry if unaffected.
Resequencing of 109 genes under the linkage peaks on chromosomes 10, 11, and 18 in 20 members of the extended kindred identified 5030 variants. From these variants, 16 SNPs in 6 genes were selected because, for each, one allele occurred primarily in either the 15 affected or the 5 unaffected kindred members; only 2 of the SNPs (rs7927241 and rs11215474) associated with VT in a larger family sample (n = 62); both are SNPs in CADM1.
In addition to these 2 SNPs, Table 2 lists 6 additional CADM1 SNPs that we identified upon further examination of the resequencing data and then genotyped in the complete kindred (n = 453). Of the 6 additional SNPs, only rs17564430 failed to associate with VT in the extended kindred, whereas all associated in the original kindred (Table 3). Further, separate tests by PC deficiency status found that the association was limited to PC-deficient persons; the rs6589488 association was most strongly supported.
CADM1 SNPs genotyped in the kindred
| SNP | Physical location (bp)* | Gene location | Risk/nonrisk alleles | Risk allele frequency |
|---|---|---|---|---|
| rs17564430 | 114548784 | exon 1 | G/T | 0.242 |
| rs10891802 | 114549098 | Promoter | C/A | 0.447 |
| rs11215397 | 114549286 | Promoter | G/A | 0.444 |
| rs3802858 | 114583702 | Intron 7 | C/T | 0.402 |
| rs6589488 | 114602166 | Intron 5 | A/T | 0.149 |
| rs10790068 | 114606777 | Intron 4 | C/T | 0.192 |
| rs7927241 | 114662630 | Intron 1 | G/C | 0.227 |
| rs11215474 | 114674839 | Intron 1 | G/C | 0.227 |
| SNP | Physical location (bp)* | Gene location | Risk/nonrisk alleles | Risk allele frequency |
|---|---|---|---|---|
| rs17564430 | 114548784 | exon 1 | G/T | 0.242 |
| rs10891802 | 114549098 | Promoter | C/A | 0.447 |
| rs11215397 | 114549286 | Promoter | G/A | 0.444 |
| rs3802858 | 114583702 | Intron 7 | C/T | 0.402 |
| rs6589488 | 114602166 | Intron 5 | A/T | 0.149 |
| rs10790068 | 114606777 | Intron 4 | C/T | 0.192 |
| rs7927241 | 114662630 | Intron 1 | G/C | 0.227 |
| rs11215474 | 114674839 | Intron 1 | G/C | 0.227 |
CADMI indicates cell adhesion molecule 1; and SNP, single nucleotide polymorphism.
Reference location from dbSNP Build 129.
P for association of each SNP with VT
| SNP* | P† for association with VT | |||||
|---|---|---|---|---|---|---|
| Original kindred | Extended kindred | |||||
| All | PC-deficient | Normal PC | All | PC-deficient | Normal PC | |
| rs17564430 | .0376 | .00985 | .489 | .332 | .278 | .467 |
| rs10891802 | .000223 | .0000215 | .284 | .00829 | .00643 | .342 |
| rs11215397 | .000299 | .0000614 | .345 | .0167 | .0148 | .355 |
| rs3802858 | .00100 | .0000514 | .300 | .0235 | .0110 | .291 |
| rs6589488 | .000215 | .0000141 | .499 | .00000720 | .00000100 | .444 |
| rs10790068 | .000727 | .0000352 | .580 | .000138 | .0000153 | .581 |
| rs7927241 | .000465 | .000726 | .202 | .0767 | .0710 | .326 |
| rs11215474 | .000465 | .000726 | .202 | .0767 | .0710 | .326 |
| SNP* | P† for association with VT | |||||
|---|---|---|---|---|---|---|
| Original kindred | Extended kindred | |||||
| All | PC-deficient | Normal PC | All | PC-deficient | Normal PC | |
| rs17564430 | .0376 | .00985 | .489 | .332 | .278 | .467 |
| rs10891802 | .000223 | .0000215 | .284 | .00829 | .00643 | .342 |
| rs11215397 | .000299 | .0000614 | .345 | .0167 | .0148 | .355 |
| rs3802858 | .00100 | .0000514 | .300 | .0235 | .0110 | .291 |
| rs6589488 | .000215 | .0000141 | .499 | .00000720 | .00000100 | .444 |
| rs10790068 | .000727 | .0000352 | .580 | .000138 | .0000153 | .581 |
| rs7927241 | .000465 | .000726 | .202 | .0767 | .0710 | .326 |
| rs11215474 | .000465 | .000726 | .202 | .0767 | .0710 | .326 |
PC indicates protein C; SNP, single nucleotide polymorphism; and VT, venous thrombosis.
n = 62 for rs7927241 and rs11215474; n = 450-453 for the other SNPs in the extended kindred.
From a mixture χ2 statistic with 2 df, testing both dominant and recessive effects.
VT was diagnosed in 86% of older PC-deficient kindred members who were heterozygous for rs6589488 compared with less than 20% of persons with either the TT genotype or normal PC (Table 4). Although the numbers were small, homozygosity of the A allele appeared not to increase risk further. Within normal PC kindred members, heterozygosity for rs6589488 increased risk only slightly, although again the numbers are small.
Affection status by protein C deficiency and rs6589488 genotype
| Protein C | rs6589488 genotype | Original kindred | Extended kindred | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Affected | Unaffected | Percent affected* | Affected | Unaffected | Percent affected* | ||||
| Age < 50 y | Age ≥ 50 y | Age < 50 y | Age ≥ 50 y | ||||||
| Normal | TT | 3 | 139 | 38 | 7.32 | 3 | 162 | 38 | 7.32 |
| Normal | AT | 4 | 74 | 27 | 12.90 | 4 | 76 | 27 | 12.90 |
| Normal | AA | 0 | 9 | 2 | 0.00 | 0 | 9 | 2 | 0.00 |
| Deficient | TT | 2 | 23 | 11 | 15.38 | 6 | 37 | 27 | 18.18 |
| Deficient | AT | 19 | 17 | 3 | 86.36 | 25 | 22 | 4 | 86.21 |
| Deficient | AA | 2 | 4 | 1 | 66.67 | 2 | 4 | 1 | 66.67 |
| Protein C | rs6589488 genotype | Original kindred | Extended kindred | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Affected | Unaffected | Percent affected* | Affected | Unaffected | Percent affected* | ||||
| Age < 50 y | Age ≥ 50 y | Age < 50 y | Age ≥ 50 y | ||||||
| Normal | TT | 3 | 139 | 38 | 7.32 | 3 | 162 | 38 | 7.32 |
| Normal | AT | 4 | 74 | 27 | 12.90 | 4 | 76 | 27 | 12.90 |
| Normal | AA | 0 | 9 | 2 | 0.00 | 0 | 9 | 2 | 0.00 |
| Deficient | TT | 2 | 23 | 11 | 15.38 | 6 | 37 | 27 | 18.18 |
| Deficient | AT | 19 | 17 | 3 | 86.36 | 25 | 22 | 4 | 86.21 |
| Deficient | AA | 2 | 4 | 1 | 66.67 | 2 | 4 | 1 | 66.67 |
Including unaffected only if age ≥ 50 years.
In the original kindred, despite the strong association with rs6589488, 9 thrombosis cases lacked one or both genetic risk factors (PC deficiency and the rs6589488 A allele), and 4 carriers of both genetic risk factors remained unaffected beyond the age of 50. Although the incomplete correspondence between thrombosis status and the presence of the 2 genetic risk factors may simply reflect variation in environmental exposure, the presence of an additional unlinked and yet undiscovered thrombophilia gene provides an alternate explanation. However, nonsignificant heritability upon accounting for rs6589488, estimated as 2.6% plus or minus 6.9% and 68.9% plus or minus 45.4% in the PC-deficient and normal subsets, respectively, failed to implicate additional thrombophilia genes. Alternatively, the incomplete correspondence between thrombosis status and the 2 genetic risk factors may result from incomplete linkage disequilibrium between rs6589488 and an unknown causative variant. Supporting this explanation, we estimated the proportion of rs6589488 A-carrying chromosomes that harbor the causative variant as 54.2% plus or minus 21.3%; the proportion of rs6589488 T-carrying chromosomes that harbor the causative variant estimated as 0. Therefore, we attribute the incomplete correspondence between thrombosis status and the 2 genetic risk factors to an additional causative variant that remains to be discovered, although environmental variation may also contribute.
Immunofluoresence
Endothelium, in a representative tissue section of saphenous vein, in Figure 1A demonstrates positive immunofluoresence staining for CADM1 stained with a rabbit polyclonal IgG antibody. Figure 1B shows similar positive immunofluoresence staining for CADM1 in the endothelium of a saphenous vein valve leaflet stained with a monoclonal chicken antibody. By using 2 different antibodies, we found that these images demonstrate for the first time the presence of CADM1 in endothelium. The chicken monoclonal CADM1 antibody was used for all subsequent CADM1 immunofluorescence staining.

Two different antibodies show CADM1 immunofluorescent staining in venous endothelium. (A) Saphenous vein wall endothelium stained positively (green) with rabbit polyclonal anti-CADM1 antibody. (B) Saphenous vein valvular leaflet endothelium stained positively with chicken monoclonal anti-CADM1 antibody.
Two different antibodies show CADM1 immunofluorescent staining in venous endothelium. (A) Saphenous vein wall endothelium stained positively (green) with rabbit polyclonal anti-CADM1 antibody. (B) Saphenous vein valvular leaflet endothelium stained positively with chicken monoclonal anti-CADM1 antibody.
In Figure 2A, confluent HUVECs in culture were stained for CADM1 and CD 31. In this image, CD 31 stains endothelial cell membrane and clusters at the cell-cell junctions, whereas CADM1 appears to be primarily cytoplasmic. The same confluent cells, stained only for CADM1 in Figure 2B, show CADM1 only in the cytoplasm. Thus, in contrast to the pulmonary epithelium, where CADM1 is localized at the cell-cell junctions,30 in confluent HUVECs CADM1 appears to be entirely cytoplasmic. In Figure 2C, migrating HUVECs show CADM1 present in the cytoplasm but also in lamellopodia and filopodia along the leading edge of the migrating HUVECs. In Figure 2D, both CADM1 and Phalloidin-bound actin appear to colocalize at the leading edge of the lamellopodia of the migrating cells. In Figure 2E, stained for both CADM1 and CD31, CADM1 is present in the filopodia and lamellopodia of the cell. The localized areas of high concentrations of CADM1 observed in both locations have been described previously.31 Figure 2F is an enlargement of the Lamellopodia outlined in Figure 2E and shows membrane-associated CADM1, with several of the localized areas of increased concentration of CADM1 staining involved in the formation of small filopodia at the leading edge of the lamellopodia.

Confluent HUVECs show cytoplasmic but not membrane immunofluorescent staining for CADM1 with chicken monoclonal anti-CADM1. In contrast, migrating HUVECs show both cytoplasmic and membrane staining for CADM1 in lamellopodia and filopodia at the leading edge of cell migration. (A) Confluent HUVECs stained for CADM1 (green) and CD31 (red). CD31 clearly demarcates the intercellular boundaries. (B) Confluent HUVECs stained only for CADM1 show no evidence of membrane staining at the intercellular boundaries. (C) Migrating HUVECs show CADM1 staining in the cytoplasm and at the leading edge of lamellopodial membranes (white arrows). (D) The image in panel C counterstained with Phalloidin (red) shows colocalization of actin with CADM1 (combination appears yellow) at the leading edge of lamellopodial membranes (white arrows). (E) Migrating HUVECs stained for CADM1 (green) and CD31 (red) show CADM1 staining of filopodia colocalized at the membrane surface with CD31. CADM1 staining is present along the walls and at the leading edge of the filopodia. White arrows denote filopodia and boxed area a lamellopodia. (F) The boxed area of panel E at greater magnification shows the presence of localized high-intensity staining of CADM1 at the leading edge of a lamellopodia, some of which appears to be associated with developing filopodia.
Confluent HUVECs show cytoplasmic but not membrane immunofluorescent staining for CADM1 with chicken monoclonal anti-CADM1. In contrast, migrating HUVECs show both cytoplasmic and membrane staining for CADM1 in lamellopodia and filopodia at the leading edge of cell migration. (A) Confluent HUVECs stained for CADM1 (green) and CD31 (red). CD31 clearly demarcates the intercellular boundaries. (B) Confluent HUVECs stained only for CADM1 show no evidence of membrane staining at the intercellular boundaries. (C) Migrating HUVECs show CADM1 staining in the cytoplasm and at the leading edge of lamellopodial membranes (white arrows). (D) The image in panel C counterstained with Phalloidin (red) shows colocalization of actin with CADM1 (combination appears yellow) at the leading edge of lamellopodial membranes (white arrows). (E) Migrating HUVECs stained for CADM1 (green) and CD31 (red) show CADM1 staining of filopodia colocalized at the membrane surface with CD31. CADM1 staining is present along the walls and at the leading edge of the filopodia. White arrows denote filopodia and boxed area a lamellopodia. (F) The boxed area of panel E at greater magnification shows the presence of localized high-intensity staining of CADM1 at the leading edge of a lamellopodia, some of which appears to be associated with developing filopodia.
We have also demonstrated CADM1 immunofluoresence staining in the cytoplasm of BOECs with the same chicken monoclonal antibody used in the endothelial cell experiments described previously (data not shown). In addition, by using a rabbit polyclonal anti-mouse SynCAM1 (Cadm1) antibody (Sigma-Aldrich) we demonstrated immunofluoresence staining for Cadm1 in the pulmonary epithelium as well as arterial and venous endothelium of C57 black mice (data not shown).
Endothelial cell culture and RNA expression array
BOECs cultured from 3 PC-deficient Kindred Vermont II members were compared with 27 healthy control subjects of mixed sex, age and race. Gene expression from BOECs of donors was analyzed with the Affymetrix U133A chip. Fold change for CADM1 of the 3 kindred members, relative to mean of control subjects, was 0.859, indicating a 15% decrease of CADM1. Analysis of CADM1 for family members versus controls produced P values of .028 (Welch t test) and .029 (SAM). Despite the small sample size, detection of a small P value is promising preliminary phenotypic data.
Discussion
This analysis has identified CADM1 as a probable thrombophilia gene that increases the risk of VT in PC-deficient kindred members. The CADM1 gene encodes an immunoglobulin-cell adhesion molecule that is involved in Ca2+- and Mg2+-independent homophilic and heterophilic binding interactions supporting intercellular adhesion.32 The cytoplasmic domain of CADM1 contains 2 binding sites, a protein 4.1 binding motif that binds DAL131 connecting CADM1 to the actin cytoskeleton and a PDZ binding motif that binds the membrane-associated guanylate cyclase MPP3,33 which interacts with the actin cytoskeleton and likely modulates cell signaling and proliferation.34
Interaction of CADM1 with the PC system can be predicted based on current knowledge of the function of each. CADM1 is the consensus name for a gene that has been called by a variety of other names in reflection of the organ system being studied, including synapse cell adhesion molecule (SynCAM1), tumor suppressor in lung cancer 1 (TSLC1), immunoglobulin super family 4 (IGSF4), and spermatogenic immunoglobulin super family (SgIGSF).30,35,,–38 CADM1 plays a variety of different roles in cell-cell adhesion. Examples of the disparate functions of CADM1 include basolateral adhesion of pulmonary epithelial cells,30 organization of synapse formation,35 adhesion of spermatogonia to Sertoli cells,39 and adhesion of mast cells to nerve and smooth muscle cells.37,38 Decreased expression of CADM1 has been associated with non–small cell lung cancer,40 other solid tumors,41,,–44 and with infertility in homozygous deficient transgenic mice.39 Interestingly, ectopic expression of CADM1 has been associated with adult T-cell leukemia with the suggestion that it mediates binding of tumor cells to endothelium, thus facilitating tumor invasion.45,46 A search of the literature failed to identify an evaluation for the presence of CADM1 in endothelial cells other than 2 articles, where it was not observed using immunohistochemistry in the pulmonary vasculature.30,47 Thus, in other cell systems CADM1 enables cell-cell adhesion, and by extension it seems likely to play a similar role in endothelial cell–cell adhesion that is essential for normal endothelial barrier function.
Activated PC (APC) stabilizes the endothelial cytoskeleton to reduce permeability, resulting in improved endothelial barrier function. For example, paracellular gaps formed upon exposure of endothelial cells to thrombin, which potentially expose circulating blood to the subendothelial extracellular matrix, can be prevented or reversed by APC bound to the endothelial cell PC receptor.48,49 This enhancement of the endothelial barrier function appears to be mediated through cortical rearrangement of the actin/myosin cytoskeleton.
We have demonstrated that CADM1 mRNA is expressed in endothelial cells, with preliminary evidence that expression is modestly decreased in members of Kindred Vermont II compared with control subjects. In addition, we have demonstrated, for the first time, the presence of CADM1 in venous endothelium by immunofluoresence. In tissue sections of saphenous vein, CADM1 appears to be present in the endothelial cytoplasm and not localized to the intercellular junctions as observed in epithelium. Cytoplasmic localization in tissue sections also has been observed in normal mesothelium, another mesodermally derived cell line.50
CADM1 does not appear to participate in cell-cell adhesion in confluent cultured HUVECs. In contrast to confluent cells, in migrating endothelial cells CADM1 is associated with the membrane at lamellopodia and filopodia along the leading edge of cell migration and appears to colocalize with actin. Colocalization of cell membrane associated actin and CADM1 has been previously reported.31,51 The colocalization of CADM1 and actin at the leading edge of endothelial cell migration in lamellopedia and filopedia, with subsequent absence of CADM1 staining at intercellular junctions once confluence is achieved, suggests that CADM1 may be involved selectively in closing gaps between endothelial cells. The presence of localized increased concentrations of CADM1 in filopodia, which play an essential role in guiding migrating endothelial cells,30,52 supports this notion. Consequently, we hypothesize that a variant form or decreased expression of CADM1, interacting with PC deficiency, impairs APC-mediated endothelial barrier protection in response to thrombogenic stimuli like thrombin generation, thereby increasing the risk of VT in kindred members.
Alternatively, if CADM1 is not the thrombophilia gene, the causative variant probably lies within 1 MB telomeric of CADM1. This localization assumes a common ancestral source of 2 distinct haplotypes that harbor the causative variant within the original kindred on the basis of haplotypes inferred with the use of SIMWALK253,54 (http://www.genetics.ucla.edu/software/simwalk, Version 2.91) of regional linkage markers and the CADM1 SNPs. The shared portion of the haplotypes extends from CADM1 SNP rs17564430 to D11S4142, a region containing 5 hypothetical proteins, but no other definite genes. Furthermore, an apparent recombination near rs7927241 shortened the haplotype segment transmitted to descendants of a second daughter of the founding couple, which, if correct, implicates CADM1 as the thrombophilia gene and localizes the causative variant between CADM1 SNPs rs17564430 and rs7927241, a distance of approximately 114 kb that encompasses the coding region of the 300 kb CADM1 gene.
The resequenced candidate gene located closest to CADM1 at 1 MB centromeric is nicotinomide N-methyltransferase (NNMT). Souto et al55 found linkage to chromosome 11q23 and association with NNMT of homocysteine level, a risk factor for VT.56 Although VT showed neither linkage nor association in their study, their sample may have lacked sufficient statistical power or genetic heterogeneity may have obscured the evidence. However, in agreement with their results, variants in NNMT did not associate with thrombosis in our resequencing sample, and NNMT falls outside our implicated region.
In summary, SNPs within the gene CADM1 strongly associate with VT in PC-deficient members of a large kindred. We found little evidence that the remaining variation in thrombosis risk results from additional thrombophilia genes. Thus, although the CADM1 SNPs tested in this analysis do not appear to include the causative variant, it is highly likely that extended resequencing of regulatory and intronic regions, with the inclusion of additional informative family members, will identify the causative variant. The interaction of abnormal or deficient endothelial CADM1 with PC deficiency suggests a novel mechanism of impaired endothelial barrier function conferring increased thrombosis risk in inherited thrombophilia that may shed light on the poorly understood third member of the Virchow triad: the vascular wall.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank members of Kindred Vermont II for their participation, Julia Valliere (University of Vermont College of Medicine) for her excellent technical support in the laboratory, and Beth Bouchard, PhD (University of Vermont College of Medicine) for cell culture support. Valuable help from Dr Douglas Taatjes, PhD, (University of Vermont College of Medicine), and Marilyn Wadsworth (University of Vermont College of Medicine) with the immunofluoresence experiments also is acknowledged. We thank Dr Kenneth Bauer (Harvard Medical School) for helpful collaboration over many years, and Dr Joseph Dickerman (University of Vermont College of Medicine), who first identified the Vermont II kindred and has collaborated in this study over the years.
This work was supported by P01 HL46703 and R01 HD017463 from the National Institutes of Health. Resequencing services were provided by J. Craig Venter Institute under contract number N01-HV-48196 from the National Heart, Lung, and Blood Institute. Support was also provided by a Transatlantic Network for Excellence in Cardiovascular Research grant from the Leducq Foundation, Paris, France.
National Institutes of Health
Authorship
Contribution: S.J.H. analyzed data, helped design the study, and wrote the paper; I.D.B. analyzed data; P.W.C., C.D., and C.Y.V. collected data; F.R.R. participated in study design and data collection; W.T. helped design and performed the immunofluorescence experiments; R.P.H. performed experiments and wrote the paper; E.G.B. designed and supervised the project, performed experiments, and wrote the paper; and all authors edited and contributed to the manuscript.
Conflict-of-interest disclosure: A patent on the BOEC culture method and use is held by the University of Minnesota and R.P.H., but there is no current financial interest. The remaining authors declare no competing financial interests.
Correspondence: Edwin G. Bovill, S254, Courtyard at the Given Bldg, University of Vermont College of Medicine, Burlington, VT 05405; e-mail: Edwin.bovill@uvm.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal