

Abstract
One major obstacle in gene therapy is the generation of immune responses directed against transgene product. Five consecutive anti-CD3 treatments concomitant with factor VIII (FVIII) plasmid injection prevented the formation of inhibitory antibodies against FVIII and achieved persistent, therapeutic levels of FVIII gene expression in treated hemophilia A mice. Repeated plasmid gene transfer is applicable in tolerized mice without eliciting immune responses. Anti-CD3 treatment significantly depleted both CD4+ and CD8+ T cells, whereas increased transforming growth factor-β levels in plasma and the frequency of both CD4+CD25+FoxP3+ and CD4+CD25−Foxp3+ regulatory T cells in the initial few weeks after treatment. Although prior depletion of CD4+CD25+ cells did not abrogate tolerance induction, adoptive transfer of CD4+ cells from tolerized mice at 6 weeks after treatment protected recipient mice from anti-FVIII immune responses. Anti-CD3–treated mice mounted immune responses against both T-dependent and T-independent neo-antigens, indicating that anti-CD3 did not hamper the immune systems in the long term. Concomitant FVIII plasmid + anti-CD3 treatment induced long-term tolerance specific to FVIII via a mechanism involving the increase in transforming growth factor-β levels and the generation of adaptive FVIII-specific CD4+Foxp3+ regulatory T cells at the periphery. Furthermore, anti-CD3 can reduce the titers of preexisting anti-FVIII inhibitory antibodies in hemophilia A mice.
Introduction
Gene therapy protocols using viral and nonviral vectors have been devised for many diseases and used in preclinical studies. Recent studies indicate that immune responses against vectors or transgene products can become major obstacles for successful application of gene therapy.1-3
Hemophilia A, a congenital bleeding disorder caused by a monogenic defect of coagulation factor VIII (FVIII), is an ideal candidate for the successful application of somatic cell gene therapy. Previous preclinical and clinical trials demonstrated that viral and nonviral gene transfer of FVIII4-9 often results in transient gene expression in the absence of immunosuppression. It is therefore essential to develop safe and effective methods to modulate immune responses against transgene products, vectors, and/or gene-engineered cells to ensure the success of gene therapy.
Therapeutic biologic compounds have been used successfully to modulate the immune system. The development of the first humanized anti-CD3 monoclonal antibody (OKT3) spurred a spate of studies with CD3-specific antibodies for the treatment of immune-mediated diseases.10-15 Clinical trials with this monoclonal antibody included treatment of acute renal allograft rejection,16,17 autoimmune insulin-dependent diabetes,18 and psoriatic arthritis,19 and prevention of islet allograft rejection.20 The results of these trials are encouraging, and anti-CD3 treatment did not elicit major side effects.16,17 Anti-CD3 therapy was demonstrated to promote, in both transplantation21,22 and autoimmune settings,23 antigen-specific immune tolerance. In patients with recent onset autoimmune diabetes, reversion of hyperglycemia was achieved by combination therapy of anti-CD3 injections and intranasal application of proinsulin peptide, but not by either agent alone.23 It was suggested that the combination therapy induces large numbers of islet cell-specific regulatory T cells (Tregs), whereas anti-CD3 or proinsulin alone was insufficient to prime Tregs.
Experimental evidence supports the hypothesis that there are 2 subsets of Tregs that differ in specificity and effector mechanism.24 Natural Tregs emerge from the thymus as a distinct lineage, whereas adaptive Tregs are induced at the periphery from CD4+CD25− T cells under specific conditions, ie, antigenic stimulation in the presence of a particular cytokine environment or altered T-cell receptor signal transduction. Studies in mice engineered to express a Foxp3 reporter confirmed that Foxp3 is a lineage marker of Tregs and correlates with suppressor activity irrespective of CD25 expression.25-27 In particular, in an autoimmune nonobese diabetic mouse model, anti-CD3 treatment induced adaptive CD4+CD25lowFoxp3+ Tregs in the periphery that suppress T-cell immunity in a transforming growth factor-β (TGF-β)–dependent manner.28
Here we report the successful use of the anti-CD3 monoclonal antibody in modulating immune responses against FVIII after gene therapy. Five consecutive injections of anti-CD3 induced tolerance to FVIII in hemophilia A mice, manifested by persistence of FVIII activity and the absence of circulatory FVIII-specific inhibitory antibodies. The treatment with anti-CD3 caused temporary depletion of CD4+ and CD8+ T cells and increased the frequency of CD4+Foxp3+ Tregs. Long-term tolerance was confirmed by a second plasmid challenge to anti-CD3 tolerized mice without eliciting FVIII-specific immune responses. In addition, anti-CD3 antibody treatment did not damper immune response toward unrelated antigens after treated mice recovered from transient immunosuppression.
Methods
Mice
All mice were kept in accordance with National Institutes of Health guidelines for animal care and the guideline of Seattle Children's Research Institute, and maintained at a specific pathogen–free facility. The animal protocols used in this study were approved by the Institutional Animal Care and Use Committee of Seattle Children's Research Institute. Hemophilia A mice in a 129/SV × C57BL/6 mixed genetic background were generated by targeted disruption of exon 16 of the FVIII gene29 and bred in our animal facility.
Antibodies
Anti–mouse-CD3ϵ monoclonal antibodies (145-2C11), anti–mouse-CD25 monoclonal antibodies (PC61), and mouse IgG1 isotype control were purchased from BioXCell. Anti–mouse-Foxp3 (FJK-16s)–fluorescein isothiocyanate, antimouse-CD25 (PC61)–allophycocyanin, anti–mouse-CD25 (7D4)–allophycocyanin, anti–mouse-CD4 (L3T4)–phycoerythrin, and anti–mouse-CD8 (Ly3)–allophycocyanin were purchased from eBioscience. Anti-CD4 (L3T4)–AlexaFlour 700 was purchased from BD Biosciences PharMingen.
Gene transfer of FVIII into hemophilia A mice with immunomodulation regimen by anti-CD3 antibodies
Hemophilia A mice were injected with 50 μg of FVIII plasmid (pBS-HCRHP-FVIIIA) in 2 mL of phosphate-buffered saline via tail vein in 8 to 10 seconds. For immunomodulation, FVIII plasmid-treated mice were given intravenous injections of anti-CD3 antibody at a dose of 40 μg at the time of plasmid injection and subsequent daily injections for 4 additional days. Groups of anti-CD3 only–treated mice, FVIII plasmid only–treated mice, and naive mice were included as controls. Blood samples were taken from the retro-orbital plexus periodically and assessed for FVIII activity and anti-FVIII antibody levels. Selected immunomodulated mice received a second plasmid challenge at 23 weeks after the first plasmid injection or received 2.5 units (∼ 500 ng) recombinant human FVIII (Baxter) emulsified in complete Freund adjuvant (CFA; Sigma-Aldrich) at 12 weeks after the first plasmid injection.
Assays for measuring FVIII activities and anti-FVIII inhibitory antibodies
FVIII activity was measured by a modified clotting assay using reagents to measure activated partial thromboplastin time and FVIII-deficient plasma.9,30 FVIII activity was calculated from a standard curve generated using serially diluted normal human pooled plasma.
Inhibitory antibodies against FVIII were measured by Bethesda assay as previously described.31 hFVIII-specific antibodies (total IgGs) were assessed by enzyme-linked immunosorbent assay (ELISA) using the same method described previously.9 Sera collected 2 weeks before and after second FVIII plasmid challenge were used.
T-cell staining and flow cytometric analysis
Single-cell suspensions were prepared by mechanical disruption of lymph nodes (superficial cervical) and spleens of mice treated with FVIII plasmid + anti-CD3, FVIII plasmid only, or anti-CD3 only, or untreated naive control mice. Peripheral blood mononuclear cells were isolated from heparinized blood after lysis of red blood cells. Cells were first stained for surface markers CD3, CD4, CD8, and CD25, and subsequently stained intracellularly for Foxp3 following the company protocol (eBioscience). Flow cytometric analysis was performed using a FACS LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (TreeStar).
Proliferation assay and suppressive assay
CD4+ T cells were isolated from spleens of mice by magnetic activated cells sorting (MACS; Miltenyi Biotec). The CD4+CD25−, CD4+CD25+ subsets were further isolated from the CD4+ T cells using a CD25+ Treg MACS isolation kit (Miltenyi Biotec). For proliferation, 1.0 × 105 CD4+ T cells were cocultured with 1.0 × 105 CD4− cells (irradiated, used as antigen presenting cells [APCs]) per well in 96-well round-bottom plate with or without the presence of FVIII at 10 U/mL (1U = 100 ng of FVIII protein) for 72 hours, followed by adding 1 μCi of [3H]thymidine per well for the final 18 hours. [3H]Thymidine incorporation was measured as counts per minute in a Betaplate scintillation counter (PerkinElmer Life and Analytical Sciences). For suppressive assay, CD4+ T cells from spleens of FVIII plasmid only–treated mice were used as responders and CD4+CD25+ T cells from spleens of FVIII plasmid + anti-CD3–treated mice at week 2 or week 6 after plasmid injection or from spleens of age-matched naive mice were used as Tregs from treated or control mice. To the coculture of 0.8 × 105 CD4+ T cells and 1.5 × 105 APCs, we added CD4+CD25+ T cells at indicated ratios. The cultures were stimulated with 10 U/mL of FVIII for 72 hours, followed by adding 1 μCi of [3H]thymidine per well for the final 18 hours. All cultures were done in triplicate. Suppression was calculated as follows32 :
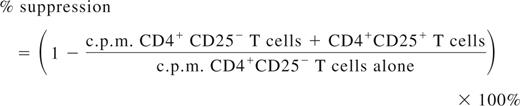
Adoptive transfer of CD4+ T cells into syngeneic hemophilia A mice
CD4+ T cells were isolated as described in the previous paragraph from spleens of FVIII plasmid + anti-CD3, FVIII plasmid only, and naive mice. Two to 4 × 106 CD4+ T cells suspended in 300 μL of phosphate-buffered saline was injected into syngeneic mice via tail vein. Mice were subsequently challenged by hydrodynamic delivery of FVIII plasmid the following day. FVIII activity and anti-FVIII inhibitory antibody levels were monitored over time.
Tregs depletion by anti-CD25 monoclonal antibodies
Hemophilia A mice (n = 4) received a single injection of 1 mg of anti-CD25 monoclonal antibodies (PC61) at 3 days before the first anti-CD3 and FVIII plasmid coinjection. Two weeks later, each mouse received an additional injection of 0.5 mg of anti-CD25. Depletion of CD4+CD25+ cells was examined by flow cytometric assay of blood samples obtained from treated mice using the 7D4 clone of anti-CD25. In an independent experiment, hemophilia A mice (n = 4), coinjected with FVIII plasmid and anti-CD3 as described in “Gene transfer of FVIII into hemophilia A mice with immunomodulation regimen by anti-CD3 antibodies,” were treated with 0.5 mg of anti-CD25 every week for 4 weeks. FVIII activities and anti-FVIII inhibitory antibodies in those mice were monitored over time.
Quantitation of systemic TGF-β levels
To quantitate systemic TGF-β1 in mice, plasma was pretreated with 1M HCL and then assayed using a TGF-β1 ELISA kit (BD Bioscience) according to the manufacturer's recommendations and the data interpolated against the linear range on the standard curves.
Immunization with the neo-T–independent antigen TNP-Ficoll
Groups of tolerized mice (n = 4) and FVIII plasmid only–treated mice (n = 3) were immunized with 20 μg of trinitrophenyl (TNP)–Ficoll intraperitoneally each 24 weeks after plasmid injection. Serum was collected10 days after TNP-Ficoll injection. Anti-TNP IgG3 and anti-TNP IgM in serum were detected by ELISA.
Immunization of mice with bacteriophage Φx174
Bacteriophage Φx174 was prepared as previously described.33 The stock solution of 1011 plaque-forming units (PFUs) per milliliter was diluted and injected intraperitoneally into 2 tolerized hemophilia A mice 6 months after plasmid injection and 2 naive hemophilia A mice at a dose of 1010 PFU/kg (2 × 108 PFU/mouse). A secondary immunization was carried out 4 weeks after the primary immunization.
Serum samples were analyzed for phage-neutralizing antibody activity and expressed as the rate of phage inactivation (Kv) using the standard formula33 : Kv = (dilution of serum/time of phage-serum incubation in minutes) × ln (phage assay PFU at 0 minute/phage assay PFU at 60 minutes). Antibody resistant to 2-mercaptoethanol was considered to be of the IgG isotype.
Statistics
Data are expressed as mean plus or minus SD. The statistical significance of the difference between means was determined using the 2-tailed Student t test. Differences were considered significant at P less than .05.
Results
Anti-CD3 induces tolerance to FVIII after FVIII plasmid-mediated gene therapy
To test whether CD3-specific antibody can modulate transgene-specific immune responses after FVIII plasmid-mediated gene therapy, we injected hemophilia A mice (n = 13/group) with 50 μg of FVIII plasmid by hydrodynamic injection (day 0) and gave 5 daily infusions of anti-CD3ϵ at a dose of 40 μg/day (days 0-4) via tail vein. Plasma samples were collected from treated and naive mice at scheduled time points, and FVIII activities and inhibitory antibody titers were assessed.
As we have previously shown,34,35 injection of FVIII plasmid alone produced short-term high-levels of FVIII activity in hemophilia A mice, followed by a gradual decrease to undetectable levels in 2 to 4 weeks because of the development of anti-FVIII inhibitory antibodies (Figure 1A-B). In contrast, immunomodulation with anti-CD3 led to persistently therapeutic levels of FVIII activities (30%-105% of FVIII levels in normal human plasma) for up to 24 weeks in 11 of the 13 FVIII plasmid-treated mice (Figure 1C). For the remaining 2 anti-CD3–treated mice, FVIII activities persisted at therapeutic levels for 10 and 15 weeks, respectively, before dropping to undetectable levels (Figure 1C). Most importantly, none of the 13 treated mice developed FVIII inhibitory antibodies throughout the 24-week experimental period (Figure 1D). In addition, when we treated mice using a lower dosage of anti-CD3 (5 μg/day at days 0-4, n = 4/group), none of the mice developed antibody response up to 12 weeks past plasmid injection, indicating that this lower dosage was also able to induce tolerance to FVIII in hemophilia A mice.

Long-term FVIII expression in hemophilia A mice after FVIII plasmid-mediated gene therapy and immunomodulation with anti-CD3. Hemophilia A mice were treated with 50 μg of FVIII plasmid by hydrodynamic injection (n = 4) or with 50 μg of FVIII plasmid at day 0 and 40 μg of anti-CD3 by daily intravenous injection at days 0 to 4 (n = 13). FVIII activities were assessed by a modified activated partial thromboplastin time assay and the anti-FVIII antibody titers by Bethesda assay over time. For plasmid only–treated mice: (A) FVIII activity and (B) anti-FVIII antibody titers. For FVIII plasmid + anti-CD3–treated mice: (C) FVIII activity and (D) anti-FVIII antibody titers. Each symbol represents data obtained from an individual mouse.
Long-term FVIII expression in hemophilia A mice after FVIII plasmid-mediated gene therapy and immunomodulation with anti-CD3. Hemophilia A mice were treated with 50 μg of FVIII plasmid by hydrodynamic injection (n = 4) or with 50 μg of FVIII plasmid at day 0 and 40 μg of anti-CD3 by daily intravenous injection at days 0 to 4 (n = 13). FVIII activities were assessed by a modified activated partial thromboplastin time assay and the anti-FVIII antibody titers by Bethesda assay over time. For plasmid only–treated mice: (A) FVIII activity and (B) anti-FVIII antibody titers. For FVIII plasmid + anti-CD3–treated mice: (C) FVIII activity and (D) anti-FVIII antibody titers. Each symbol represents data obtained from an individual mouse.
To assess transgene-specific T-cell proliferation, we isolated splenic CD4+ T cells from naive, anti-CD3 only, FVIII plasmid only, and FVIII plasmid + anti-CD3–treated mice 3 weeks after plasmid injection. These responder cells were cocultured with irradiated splenic CD4− cells from a naive mouse, which served as APCs. Without stimulant, T cells from none of the experimental groups proliferated (Figure 2). When FVIII protein was added at optimal concentration, CD4+ T cells from FVIII plasmid only–treated group proliferated robustly, as expected. In contrast, CD4+ T cells from FVIII plasmid + anti-CD3–treated mice 3 weeks (Figure 2) and 6 weeks (data not shown) after plasmid injection showed only minor nonspecific proliferation, similar to CD4+ T cells from control naive and anti-CD3 only–treated mice. These results are consistent with the observations that anti-CD3 treatment effectively suppressed transgene-specific immune responses after gene transfer of FVIII plasmid.
![Figure 2. Absence of recall proliferation in CD4+ T cells from FVIII plasmid plus anti-CD3–treated mice. CD4+ T cells were isolated by MACS from spleens of naive, anti-CD3 only, FVIII plasmid only, and FVIII plasmid + anti-CD3–treated mice 3 weeks after plasmid injection. A total of 1.0 × 105 CD4+ T cells were cocultured with irradiated 1.0 × 105 CD4− cells in 96-well round-bottom plates with or without the presence of FVIII at 10 U/mL for 72 hours, followed by adding 1 μCi of [3H]thymidine per well for the final 18 hours. *P < .05, compared with the group of FVIII plasmid only. Data shown are mean ± SD of counts per minute of [3H]thymidine incorporation in triplicate wells.](/view-large/figure/7976098/zh89990944690002.jpeg)
Absence of recall proliferation in CD4+ T cells from FVIII plasmid plus anti-CD3–treated mice. CD4+ T cells were isolated by MACS from spleens of naive, anti-CD3 only, FVIII plasmid only, and FVIII plasmid + anti-CD3–treated mice 3 weeks after plasmid injection. A total of 1.0 × 105 CD4+ T cells were cocultured with irradiated 1.0 × 105 CD4− cells in 96-well round-bottom plates with or without the presence of FVIII at 10 U/mL for 72 hours, followed by adding 1 μCi of [3H]thymidine per well for the final 18 hours. *P < .05, compared with the group of FVIII plasmid only. Data shown are mean ± SD of counts per minute of [3H]thymidine incorporation in triplicate wells.
Absence of recall proliferation in CD4+ T cells from FVIII plasmid plus anti-CD3–treated mice. CD4+ T cells were isolated by MACS from spleens of naive, anti-CD3 only, FVIII plasmid only, and FVIII plasmid + anti-CD3–treated mice 3 weeks after plasmid injection. A total of 1.0 × 105 CD4+ T cells were cocultured with irradiated 1.0 × 105 CD4− cells in 96-well round-bottom plates with or without the presence of FVIII at 10 U/mL for 72 hours, followed by adding 1 μCi of [3H]thymidine per well for the final 18 hours. *P < .05, compared with the group of FVIII plasmid only. Data shown are mean ± SD of counts per minute of [3H]thymidine incorporation in triplicate wells.
To further investigate whether anti-CD3 immunomodulation induced short-term unresponsiveness or long-term tolerance to FVIII, we challenge the anti-CD3 immunomodulated mice a second time at 23 weeks after the first FVIII plasmid injection. For each independent experiment (n = 3 mice/group, total 2 groups, representative data from one group were shown in Figure 3), we chose 2 mice in which long-term FVIII expression had been established and one mouse in which FVIII expression fell to undetectable levels within 12 weeks after treatment. The second challenge with FVIII plasmid induced a short burst of high-level FVIII activities in all 3 mice (Figure 3A). Mice with persistent FVIII expression after the first FVIII plasmid injection continued to generate detectable FVIII expression after the second challenge (Figure 3A). In contrast, the mouse with undetectable FVIII expression 12 weeks after the first treatment only boosted FVIII expression for a couple of weeks after the second plasmid challenge then became again undetectable (Figure 3A). None of the anti-CD3–treated mice developed detectable inhibitory antibodies against FVIII after the second challenge (Figure 3B). In an independent control experiment, we challenged FVIII plasmid only–treated mice with a second FVIII plasmid injection at 8 weeks. In contrast to mice treated with anti-CD3, none of the FVIII plasmid only–treated mice produced a boost in FVIII activity, but all showed a significant increase in anti-FVIII antibody after second challenge.

Maintenance of immune tolerance to FVIII after a second challenge with FVIII plasmid. FVIII plasmid + anti-CD3–treated mice were given a second plasmid challenge at 23 weeks after the first plasmid injection. Three mice were chosen: 2 of these had persistent FVIII activity and the other one had lost FVIII activity. (A) FVIII activity and (B) anti-FVIII antibody titers were examined as described in Figure 1. Each line represents an individual mouse. Data shown are from one representative experiment (2 independent experiments, n = 3/group).
Maintenance of immune tolerance to FVIII after a second challenge with FVIII plasmid. FVIII plasmid + anti-CD3–treated mice were given a second plasmid challenge at 23 weeks after the first plasmid injection. Three mice were chosen: 2 of these had persistent FVIII activity and the other one had lost FVIII activity. (A) FVIII activity and (B) anti-FVIII antibody titers were examined as described in Figure 1. Each line represents an individual mouse. Data shown are from one representative experiment (2 independent experiments, n = 3/group).
Next, we assessed the total human FVIII-specific antibodies (total IgGs) in FVIII plasmid only mice, FVIII plasmid + anti-CD3 mice of short-term FVIII expression or long-term expression (n = 2/group) before and after second plasmid challenge. We detected high levels of FVIII-specific antibodies (∼ 27 μg/mL) in FVIII plasmid only mice but not in FVIII plasmid + anti-CD3 mice of long-term FVIII expression before and after the second plasmid challenge (supplemental Table 1, available on the Blood website; see the Supplemental Materials link at the top of the online article). However, we detected low levels of nonneutralizing antibodies (∼ 70 ng/mL) in FVIII plasmid + anti-CD3 mice of short-term FVIII expression at 2 weeks before and after second plasmid injection (supplemental Table 1), which may be responsible for eliminating circulating FVIII protein in mouse plasma.
Furthermore, we challenged 2 groups of mice (FVIII plasmid only and FVIII plasmid + anti-CD3 tolerized) with 2.5 units of recombinant hFVIII emulsified in CFA at 12 weeks after FVIII plasmid injection. Two weeks after the second challenge, we found that FVIII expression persisted at the same level as that before the challenge, and no inhibitor antibody response was detected in previously FVIII plasmid + anti-CD3 tolerized mice (data not shown). In contrast, CFA + FVIII protein challenge induced a robust secondary immune response in FVIII plasmid only–treated mice. FVIII plasmid + anti-CD3–treated mice were tolerant to hFVIII protein even when presented in stringent conditions. Taken together, these data indicate that anti-CD3 treatment exerted long-term partial or complete protection against FVIII-specific immune responses after FVIII plasmid-mediated gene therapy in hemophilia A mice.
Anti-CD3 treatment induces depletion of CD4+ and CD8+ T cells and an increase in the frequency of CD4+Foxp3+ T cell
Because anti-CD3 treatment targets the T-cell receptor complex, we investigated the T-cell compartments of mice injected with anti-CD3. We analyzed spleen, lymph node, and peripheral blood in groups of mice (n = 3/group) treated with anti-CD3 only, FVIII plasmid only, or FVIII plasmid + anti-CD3 and naive control mice. On day 3 after the last of 5 anti-CD3 injections (day 8 after FVIII plasmid injection), approximately 80% to 90% of CD4+ and CD8+ T cells were depleted in spleen, lymph nodes, and blood of anti-CD3 only and FVIII plasmid + anti-CD3–treated mice but not in plasmid only–treated animals (Figure 4A,D). A detailed flow-cytometric analysis of peripheral blood lymphocytes showed that the levels of CD4+ T cells declined most significantly at week 1 and gradually returned to normal within 8 weeks in both anti-CD3 only and FVIII plasmid + anti-CD3–treated groups (Figure 4D). FVIII plasmid injection had no effect on anti-CD3–induced CD4+ T-cell depletion.

Effect of anti-CD3 treatment on CD4+, CD8+ T cells, and CD4+Foxp3+ Tregs in spleen and blood. (A-C) Spleen cells were isolated from FVIII plasmid only and FVIII plasmid + anti-CD3–treated mice (n = 3/group). (A) Cells were stained and analyzed for CD4+, CD8+, and CD4+CD25+Foxp3+ T cells by flow cytometry 8 days after the first anti-CD3 injection and FVIII plasmid transfer. (Left panel) Representative dot plots. (Right panel) Summary of the data from 2 groups of mice. (B) Representative histograms of CD3 expression on CD4+Foxp3+ and CD4+Foxp3− T cells obtained from FVIII plasmid only– (left panel) and FVIII plasmid + anti-CD3 (right panel)–treated mice. Dark line represents CD4+Foxp3− cells; light line, CD4+Foxp3+ cells. (C) Detailed analysis of CD25 expression on CD4+Foxp3+ Tregs at day 8, week 4, and week 6 after treatment. (Top panel) Representative dot plots of Foxp3+ expression on CD4+ T cells (CD4+Foxp3+ cells are gated). (Bottom panel) Representative dot plots of CD25+ expression on CD4+Foxp3+ T cells (CD4+CD25hiFoxp3+, CD4+CD25lowFoxp3+, and CD4+CD25−Foxp3+ are each gated). Numbers are percentages of corresponding populations. Data shown are mean ± SD. **P < .01. (D-F) Blood samples were collected at serial time points from the 4 groups, including naive untreated, anti-CD3 only, FVIII plasmid only, and FVIII plasmid + anti-CD3–treated mice, stained and analyzed by flow cytometry for CD4+ T cells (D), CD4+Foxp3+ Tregs (E), and CD4+CD25+Foxp3+ Tregs (F). Data shown are mean ± SD. **P < .01 compared with FVIII plasmid only. #P < .05 compared with anti-CD3 only.
Effect of anti-CD3 treatment on CD4+, CD8+ T cells, and CD4+Foxp3+ Tregs in spleen and blood. (A-C) Spleen cells were isolated from FVIII plasmid only and FVIII plasmid + anti-CD3–treated mice (n = 3/group). (A) Cells were stained and analyzed for CD4+, CD8+, and CD4+CD25+Foxp3+ T cells by flow cytometry 8 days after the first anti-CD3 injection and FVIII plasmid transfer. (Left panel) Representative dot plots. (Right panel) Summary of the data from 2 groups of mice. (B) Representative histograms of CD3 expression on CD4+Foxp3+ and CD4+Foxp3− T cells obtained from FVIII plasmid only– (left panel) and FVIII plasmid + anti-CD3 (right panel)–treated mice. Dark line represents CD4+Foxp3− cells; light line, CD4+Foxp3+ cells. (C) Detailed analysis of CD25 expression on CD4+Foxp3+ Tregs at day 8, week 4, and week 6 after treatment. (Top panel) Representative dot plots of Foxp3+ expression on CD4+ T cells (CD4+Foxp3+ cells are gated). (Bottom panel) Representative dot plots of CD25+ expression on CD4+Foxp3+ T cells (CD4+CD25hiFoxp3+, CD4+CD25lowFoxp3+, and CD4+CD25−Foxp3+ are each gated). Numbers are percentages of corresponding populations. Data shown are mean ± SD. **P < .01. (D-F) Blood samples were collected at serial time points from the 4 groups, including naive untreated, anti-CD3 only, FVIII plasmid only, and FVIII plasmid + anti-CD3–treated mice, stained and analyzed by flow cytometry for CD4+ T cells (D), CD4+Foxp3+ Tregs (E), and CD4+CD25+Foxp3+ Tregs (F). Data shown are mean ± SD. **P < .01 compared with FVIII plasmid only. #P < .05 compared with anti-CD3 only.
Anti-CD3 treatment resulted in a significantly higher percentage of CD4+Foxp3+ T cells in spleen (Figure 4A), lymph node (supplemental Figure 1), and peripheral blood (Figure 4E) in mice. The absolute number of splenic CD4+FoxP3+ T cells in FVIII plasmid + anti-CD3–treated mice were approximately 90% of that observed in FVIII plasmid only–treated mice based on total number of spleen cells and flow analysis (n = 3, P = .37; Table 1). Consistent with this observation is our finding that CD3 expression is higher on the surface of splenic CD4+Foxp3− T cells (median fluorescence intensity = 6147 ± 217, Figure 4B left panel, dark line) than on splenic CD4+Foxp3+ T cells (median fluorescence intensity = 4086 ± 42, Figure 4B left panel, thin line) in plasmid FVIII only–treated mice. With anti-CD3 treatment, the CD3high subsets in both populations of CD4+Foxp3− and CD4+Foxp3+ were depleted (Figure 4B right panel). Anti-CD3 treatment selectively depleted more CD4+Foxp3− T cells than CD4+Foxp3+ T cells.
Absolute numbers of distinct populations of CD4+ T cells from FVIII plasmid + anti-CD3– and FVIII plasmid only–treated mice at day 8, week 4, and week 6 after treatment
| Treatment group/cell type | Day 8 | Week 4 | Week 6 |
|---|---|---|---|
| FVIII plasmid only | |||
| CD4+ cells | 10.8 (± 2.5) × 106 | 11.0 (± 1.8) × 106 | 11.4 (± 1.0) × 106 |
| CD4+Foxp3+ cells | 1.43 (± 0.20) × 106 | 1.32 (± 0.30) × 106 | 1.35 (± 0.30) × 106 |
| CD4+CD25+Foxp3+ cells | 1.1 (± 0.09) × 106 | 1.0 (± 0.24) × 106 | 1.2 (± 0.1) × 106 |
| CD4+CD25−Foxp3+ cells | 1.7 (± 0.37) × 105 | 1.9 (± 0.03) × 105 | 1.8 (± 0.23) × 105 |
| FVIII plasmid + anti-CD3 | |||
| CD4+ cells | 4.2 (± 1.1) × 106* | 7.7 (± 0.8) × 106* | 10.4 (± 1.5) × 106 |
| CD4+Foxp3+ cells | 1.28 (± 0.16) × 106 | 1.38 (± 0.13) × 106 | 1.4 (± 0.1) × 106 |
| CD4+CD25+Foxp3+ cells | 0.74 (± 0.17) × 106* | 1.0 (± 0.1) × 106 | 1.1 (± 0.08) × 106 |
| CD4+CD25−Foxp3+ cells | 5.6 (± 1.0) × 105† | 2.1 (± 0.02) × 105 | 1.9 (± 0.13) × 105 |
| Treatment group/cell type | Day 8 | Week 4 | Week 6 |
|---|---|---|---|
| FVIII plasmid only | |||
| CD4+ cells | 10.8 (± 2.5) × 106 | 11.0 (± 1.8) × 106 | 11.4 (± 1.0) × 106 |
| CD4+Foxp3+ cells | 1.43 (± 0.20) × 106 | 1.32 (± 0.30) × 106 | 1.35 (± 0.30) × 106 |
| CD4+CD25+Foxp3+ cells | 1.1 (± 0.09) × 106 | 1.0 (± 0.24) × 106 | 1.2 (± 0.1) × 106 |
| CD4+CD25−Foxp3+ cells | 1.7 (± 0.37) × 105 | 1.9 (± 0.03) × 105 | 1.8 (± 0.23) × 105 |
| FVIII plasmid + anti-CD3 | |||
| CD4+ cells | 4.2 (± 1.1) × 106* | 7.7 (± 0.8) × 106* | 10.4 (± 1.5) × 106 |
| CD4+Foxp3+ cells | 1.28 (± 0.16) × 106 | 1.38 (± 0.13) × 106 | 1.4 (± 0.1) × 106 |
| CD4+CD25+Foxp3+ cells | 0.74 (± 0.17) × 106* | 1.0 (± 0.1) × 106 | 1.1 (± 0.08) × 106 |
| CD4+CD25−Foxp3+ cells | 5.6 (± 1.0) × 105† | 2.1 (± 0.02) × 105 | 1.9 (± 0.13) × 105 |
P < .05 compared with data of FVIII plasmid only.
P < .01 compared with data of FVIII plasmid only.
Furthermore, after anti-CD3 treatment, the frequency of CD4+CD25+Foxp3+ T cells increased significantly in the peripheral blood with or without concomitant FVIII plasmid injection (Figure 4F). Although there was no difference in the percentage of CD4+CD25+Foxp3+ T cells between mice treated with anti-CD3 only and FVIII plasmid + anti-CD3, there was a significant difference in the percentage of CD4+Foxp3+ T cells between these 2 groups of mice (Figure 4E, P < .05), indicating that antigen exposure promoted generation of CD4+Foxp3+ T cells. Similar results were observed in the CD4+ T-cell compartments of spleen (Figure 4A) and lymph node (supplemental Figure 1). Despite the percentage of CD25+Foxp3+ Tregs in CD4+ T cells increased significantly, the absolute number of splenic CD4+CD25+Foxp3+ Tregs in FVIII plasmid + anti-CD3–treated mice were approximately 67% of that observed in FVIII plasmid only–treated mice at day 3 (n = 3, P < .05; Table 1).
A close examination of CD25 expression on CD4+Foxp3+ T cells revealed that approximately half of the CD4+Foxp3+ T cells is CD25− and only a small fraction of CD4+Foxp3+ T cells is CD25hi in spleens (Figure 4C) and lymph nodes (supplemental Figure 1) of mice treated with FVIII plasmid + anti-CD3 at day 8 after FVIII plasmid injection. In comparison, approximately half of the CD4+Foxp3+ T cells is CD25hi and only a small fraction of the CD4+Foxp3+ T cells are CD25− in spleens and lymph nodes of naive mice and FVIII plasmid only–treated mice (Figure 4C; supplemental Figure 1). At 4 and 6 weeks after plasmid injection, the distribution of CD25+ and CD25− cells in CD4+Foxp3+ T cells in spleens of FVIII plasmid + anti-CD3 mice gradually returned to levels comparable with that in spleens of naive mice and FVIII plasmid only–treated mice (Figure 4C).
A recent study suggested that CD3-specific antibody increases the level of TGF-β1 in vivo, leading to the generation of CD4+CD25+Foxp3+ regulatory T cells and tolerance.36 We investigated the TGF-β1 levels in the plasma of naive and FVIII plasmid + anti-CD3–treated mice. Two FVIII plasmid + anti-CD3 tolerized mice (mouse 4 and mouse 5, Table 2) had increased TGF-β1 levels at day 3 and/or day 10 after the last anti-CD3 injection, compared with that of naive mice (mice 1, 2, and 3). Mouse 6, which lost FVIII activity at approximately 12 weeks, did not produce higher plasma TGF-β1 at day 3 and/or day 10.
Anti-CD3 treatment increases systemic TGF-β1
| Mouse no. | Systemic TGF-β1 (plasma, ng/mL) |
|---|---|
| Naive mice | |
| 1 | 15.2 ± 0.99 |
| 2 | 10.7 ± 1.3 |
| 3 | 8.6 ± 1.4 |
| FVIII plasmid + anti-CD3 mice | |
| 4, day 3 | 43.25 ± 2.28 |
| 4, day 10 | 39.97 ± 0.27 |
| 5, day 3 | 19.35 ± 0.32 |
| 5, day 10 | 30.97 ± 1.45 |
| 6, day 3 | 7.56 ± 0.35 |
| 6, day 10 | 15.44 ± 0.66 |
| Mouse no. | Systemic TGF-β1 (plasma, ng/mL) |
|---|---|
| Naive mice | |
| 1 | 15.2 ± 0.99 |
| 2 | 10.7 ± 1.3 |
| 3 | 8.6 ± 1.4 |
| FVIII plasmid + anti-CD3 mice | |
| 4, day 3 | 43.25 ± 2.28 |
| 4, day 10 | 39.97 ± 0.27 |
| 5, day 3 | 19.35 ± 0.32 |
| 5, day 10 | 30.97 ± 1.45 |
| 6, day 3 | 7.56 ± 0.35 |
| 6, day 10 | 15.44 ± 0.66 |
Systemic TGF-β1 in the plasma was quantified by ELISA with duplicates. Day 3 and day 10 indicate days after the last anti-CD3 injection. Data are mean ± SD of individual mice.
Examination of the role of CD4+CD25+Foxp3+ Tregs in tolerance induction by anti-CD3 treatment
Because Tregs have been involved in the induction of peripheral tolerance in many settings, we tested a possible role of Tregs in our model. We first examined the suppressive activity of CD4+CD25+ T cells isolated from FVIII plasmid + anti-CD3 tolerized and from naive mice at 2 weeks after FVIII plasmid injection in a FVIII-specific suppression assay. CD4+ T cells from FVIII plasmid only–treated mice were used as responder T cells. CD4+CD25+ Tregs of naive mice showed no suppression on FVIII-specific proliferation. Surprisingly, CD4+CD25+ Tregs of FVIII plasmid + anti-CD3 mice, collected 2 weeks after FVIII plasmid injection, did not suppress FVIII-specific proliferation (Figure 5A). However, when we used CD4+CD25+Treg cells isolated from tolerized mice 6 weeks or more after FVIII plasmid injection, we observed FVIII-specific suppression (Figure 5B).
![Figure 5. FVIII-specific suppressive capability of CD4+CD25+ T cells derived from FVIII plasmid + anti-CD3–treated mice. For the FVIII-specific suppressive assay, we used as responder CD4+ T (Tresp) cells from the spleen of a FVIII plasmid only–treated mouse at week 4 after plasmid injection. CD4+CD25+ T cells from pooled splenic cells of naive hemophilia A mice or FVIII plasmid + anti-CD3–treated mice were used as suppressive cells. The final coculture system consisted of 0.8 × 105 CD4+ Tresp cells, 1.5 × 105 irradiated CD4− cells, and CD4+CD25+ Treg cells at the indicated Treg/Tresp ratio. (A) CD4+CD25+ Tregs were isolated at week 2 after plasmid injection from mice treated with FVIII plasmid + anti-CD3. (B) CD4+CD25+ Tregs were isolated at week 6 after plasmid injection from mice treated with FVIII plasmid + anti-CD3. Data shown are mean ± SD of counts per minute of [3H]thymidine incorporation in triplicate wells.](/view-large/figure/7976114/zh89990944690005.jpeg)
FVIII-specific suppressive capability of CD4+CD25+ T cells derived from FVIII plasmid + anti-CD3–treated mice. For the FVIII-specific suppressive assay, we used as responder CD4+ T (Tresp) cells from the spleen of a FVIII plasmid only–treated mouse at week 4 after plasmid injection. CD4+CD25+ T cells from pooled splenic cells of naive hemophilia A mice or FVIII plasmid + anti-CD3–treated mice were used as suppressive cells. The final coculture system consisted of 0.8 × 105 CD4+ Tresp cells, 1.5 × 105 irradiated CD4− cells, and CD4+CD25+ Treg cells at the indicated Treg/Tresp ratio. (A) CD4+CD25+ Tregs were isolated at week 2 after plasmid injection from mice treated with FVIII plasmid + anti-CD3. (B) CD4+CD25+ Tregs were isolated at week 6 after plasmid injection from mice treated with FVIII plasmid + anti-CD3. Data shown are mean ± SD of counts per minute of [3H]thymidine incorporation in triplicate wells.
FVIII-specific suppressive capability of CD4+CD25+ T cells derived from FVIII plasmid + anti-CD3–treated mice. For the FVIII-specific suppressive assay, we used as responder CD4+ T (Tresp) cells from the spleen of a FVIII plasmid only–treated mouse at week 4 after plasmid injection. CD4+CD25+ T cells from pooled splenic cells of naive hemophilia A mice or FVIII plasmid + anti-CD3–treated mice were used as suppressive cells. The final coculture system consisted of 0.8 × 105 CD4+ Tresp cells, 1.5 × 105 irradiated CD4− cells, and CD4+CD25+ Treg cells at the indicated Treg/Tresp ratio. (A) CD4+CD25+ Tregs were isolated at week 2 after plasmid injection from mice treated with FVIII plasmid + anti-CD3. (B) CD4+CD25+ Tregs were isolated at week 6 after plasmid injection from mice treated with FVIII plasmid + anti-CD3. Data shown are mean ± SD of counts per minute of [3H]thymidine incorporation in triplicate wells.
Next, we performed adoptive transfer experiments. Pooled CD4+ T cells were isolated from the spleens of naive mice, FVIII plasmid only, and FVIII plasmid + anti-CD3–treated mice 2 or 6 weeks after plasmid injection. Two to 4 × 106 CD4+ cells were transferred into naive syngeneic hemophilia A mice. The recipient mice were challenged with FVIII plasmid 1 day after cell transfer, and FVIII expression levels and inhibitory antibodies production were monitored. As expected, transfer of CD4+ T cells of naive mice did not affect FVIII expression and formation of FVIII inhibitors in recipient mice after FVIII plasmid challenge (Table 3). Transfer of CD4+ T cells obtained from FVIII plasmid only–treated mice 2 weeks after plasmid injection accelerated the loss of FVIII activity and the formation of inhibitory antibodies in recipient mice. Interestingly, transfer of CD4+ T cells derived from FVIII plasmid + anti-CD3–treated mice 2 weeks after plasmid injection affects neither the expression of FVIII nor the formation of FVIII inhibitors in recipient mice, whereas transfer of CD4+ T cells derived from FVIII plasmid + anti-CD3–treated mice 6 weeks after plasmid injection prolonged the expression of FVIII and delayed the formation of FVIII inhibitors in recipient mice, compared with transfer of CD4+ T cells derived from naive mice.
Adoptive transfer of CD4+ cells from tolerized mice at week 6 rendered partial dominant tolerance to sygeneic mice
| CD4+ cell donors | hFVIII activity of receivers, percentage of normal | Anti-hFVIII antibodies of receivers, Bethesda units | ||||
|---|---|---|---|---|---|---|
| Day 7 | Day 14 | Day 21 | Day 7 | Day 14 | Day 21 | |
| Naive | 317.0 ± 76.2 | 2.2 ± 2.3 | 0.5 ± 0.6 | 0 | 0 | 28.5 ± 5 |
| FVIII plasmid only | 28.1 | 0 | 0 | 1.5 | 55 | 97 |
| FVIII plasmid + anti-CD3 (week 2) | 331.1 | 12.5 | 0 | 0 | 5 | 20 |
| FVIII plasmid + anti-CD3 (week 6) | 262.5 ± 9.3 | 95.2 ± 46.4 | 40.0 ± 39.7 | 0 | 0 | 0 |
| CD4+ cell donors | hFVIII activity of receivers, percentage of normal | Anti-hFVIII antibodies of receivers, Bethesda units | ||||
|---|---|---|---|---|---|---|
| Day 7 | Day 14 | Day 21 | Day 7 | Day 14 | Day 21 | |
| Naive | 317.0 ± 76.2 | 2.2 ± 2.3 | 0.5 ± 0.6 | 0 | 0 | 28.5 ± 5 |
| FVIII plasmid only | 28.1 | 0 | 0 | 1.5 | 55 | 97 |
| FVIII plasmid + anti-CD3 (week 2) | 331.1 | 12.5 | 0 | 0 | 5 | 20 |
| FVIII plasmid + anti-CD3 (week 6) | 262.5 ± 9.3 | 95.2 ± 46.4 | 40.0 ± 39.7 | 0 | 0 | 0 |
Cells were isolated from donors 2 weeks or 6 weeks after plasmid injection.
To deplete CD4+CD25+ Tregs, we treated hemophilia A mice with anti-CD25 (clone PC61) monoclonal antibodies before FVIII plasmid and anti-CD3 injection as outlined in Figure 5A. Injection of 1 mg of anti-CD25 at day 0 plus a second injection of 0.5 mg of anti-CD25 at day 14 (Figure 6A) into hemophilia A mice depleted 99% of CD4+CD25+ T cells (including both CD4+CD25+Foxp3+ and CD4+CD25+Foxp3− T cells) for up to 4 weeks but, however, did not affect CD4+CD25−Foxp3+ T cells (Figure 6B-C). The percentage of CD4+CD25−Foxp3+ T cells in anti-CD25 + FVIII plasmid + anti-CD3–treated mice was significantly higher than that observed in naive mice (P < .05; Figure 6B-C). After anti-CD25 + FVIII plasmid + anti-CD3 treatment, 2 of 4 mice lost FVIII expression at 8 to 12 weeks after plasmid injection, whereas the other 2 mice achieved persistent FVIII expression for 112 days (experimental duration). Similar results (2 of 4 mice lost expression) were observed in the control IgG1 + FVIII plasmid + anti-CD3–treated mice. None of the mice developed inhibitory antibodies during the time of study (data not shown). These results are in agreement with a previous report,14 suggesting that prior treatment with anti-CD25 does not abrogate anti-CD3 induced tolerance.

Effect of prior depletion of CD4+CD25+ T cells by anti-CD25 antibody on tolerance induction. (A) Diagram of anti-CD25 dosing schedule. (B-C) Anti-CD25 treatment depleted CD4+CD25+ T cells but not CD4+CD25−FoxP3+ T cells in peripheral blood at day 8 after plasmid injection. (B) Representative dot plots of staining. (C) Summary of the data. (D) FVIII activities in hemophilia A mice treated with anti-CD25+FVIII plasmid + anti-CD3 (n = 4/group) or with IgG1 + FVIII plasmid + anti-CD3 control (n = 4/group). One of 2 independent experiments is shown.
Effect of prior depletion of CD4+CD25+ T cells by anti-CD25 antibody on tolerance induction. (A) Diagram of anti-CD25 dosing schedule. (B-C) Anti-CD25 treatment depleted CD4+CD25+ T cells but not CD4+CD25−FoxP3+ T cells in peripheral blood at day 8 after plasmid injection. (B) Representative dot plots of staining. (C) Summary of the data. (D) FVIII activities in hemophilia A mice treated with anti-CD25+FVIII plasmid + anti-CD3 (n = 4/group) or with IgG1 + FVIII plasmid + anti-CD3 control (n = 4/group). One of 2 independent experiments is shown.
Unrelated T-dependent and T-independent antigen challenge
Because anti-CD3 treatment temporarily depleted a large portion of the T-cell compartment, the long-term effect on the immune system was investigated. Two FVIII plasmid + anti-CD3 tolerized mice and 2 control naive mice were inoculated with the T-dependent antigen, bacteriophage Φx174,33 and another 2 groups with the T-independent antigen, TNP-Ficoll, 24 weeks after plasmid delivery. Peak antibody titers observed at 2 weeks after primary and 1 week after secondary Φx174 immunization and isotype switching (percentage phage-specific IgG) during secondary immunization were not different between the 2 groups (Figure 7A). As shown in Figure 7, no significant difference in serum levels of anti-TNP IgG3 (Figure 7B) or anti-TNP IgM (Figure 7C) were found between tolerized mice and FVIII plasmid only–treated mice. Together, these data indicated that transient immunomodulation with anti-CD3 does not adversely affect immune responses to T-dependent or T-independent neoantigens.

Challenge of tolerized mice with unrelated T-dependent and T-independent antigens. Tolerized mice (n = 2) or naive mice (n = 2) were challenged with the T-dependent antigen, bacteriophage Φx174 twice 4 weeks apart starting at week 24 after plasmid injection; antibody titers, expressed as Kv, were monitored as described in “Immunization of mice with bacteriophage Φx174.” (A) Kv value over time after primary and secondary bacteriophage Φx174 immunization. The proportion of phage-neutralizing antibody of the IgG isotype was measured 2 weeks after secondary immunization by treating with 2-mercaptoethanol and is provided in panel A. We used TNP-Ficoll (20 μg) as T-independent antigen to immunize tolerized (n = 4) or FVIII plasmid only–treated hemophilia A mice (n = 3) at week 24 after plasmid injection. Serum was collected at day 10 after challenge and assessed for (B) anti-TNP IgM, and (C) anti-TNP IgG3. No significant differences were found between the groups for Kv, anti-TNP IgM, and anti-TNP IgG3 levels. Data are mean ± SD.
Challenge of tolerized mice with unrelated T-dependent and T-independent antigens. Tolerized mice (n = 2) or naive mice (n = 2) were challenged with the T-dependent antigen, bacteriophage Φx174 twice 4 weeks apart starting at week 24 after plasmid injection; antibody titers, expressed as Kv, were monitored as described in “Immunization of mice with bacteriophage Φx174.” (A) Kv value over time after primary and secondary bacteriophage Φx174 immunization. The proportion of phage-neutralizing antibody of the IgG isotype was measured 2 weeks after secondary immunization by treating with 2-mercaptoethanol and is provided in panel A. We used TNP-Ficoll (20 μg) as T-independent antigen to immunize tolerized (n = 4) or FVIII plasmid only–treated hemophilia A mice (n = 3) at week 24 after plasmid injection. Serum was collected at day 10 after challenge and assessed for (B) anti-TNP IgM, and (C) anti-TNP IgG3. No significant differences were found between the groups for Kv, anti-TNP IgM, and anti-TNP IgG3 levels. Data are mean ± SD.
Anti-CD3 can reduce the titers of preexisting inhibitory antibodies in hemophilia A mice
To date, there are no clinical or laboratory risk factors that allow the identification of a patient who will develop antibody. Therefore, it is very important to develop a strategy to treat preexisting inhibitory antibodies. Thus, we injected a group of hemophilia A mice (n = 4/group) with 50 μg FVIII plasmid by hydrodynamic injection (week −8). Eight weeks after plasmid injection, 2 mice developed high-titer anti-FVIII antibody and 2 mice developed low-titer antibody. All 4 mice were subjected to 5 daily infusions of anti-CD3ϵ at a dose of 40 μg/day (days 0-4) via tail vein. Anti-FVIII antibody titers were significantly reduced or eliminated and FVIII activities recovered to detectable levels 2 weeks after anti-CD3 treatment (Table 4). However, FVIII activities declined and anti-FVIII antibody titers reappeared at low titers or increased at later time points. These data indicated that anti-CD3 can partially modulate preexisting anti-FVIII immune responses.
Mice (n = 4) injected with 50 μg of FVIII plasmid to induce anti-FVIII inhibitors
| Week after FVIII plasmid injection | Week after anti-CD3 treatment* | ||||
|---|---|---|---|---|---|
| 1 | 4 | 8 | 2 | 4 | |
| FVIII activities, percentage of normal | |||||
| P1 | 189 | 0 | 0 | 13.3 | 11.1 |
| P2 | 258 | 0 | 0 | 155.4 | 20.5 |
| P3 | 227 | 0 | 0 | 0 | 0 |
| P4 | 304 | 0 | 0 | Died | Died |
| Anti-FVIII inhibitors, BU | |||||
| P1 | 0 | 0.3 | 0.4 | 0 | 0 |
| P2 | 0 | 50 | 62 | 0 | 3 |
| P3 | 0 | 0.3 | 1 | 0 | 0.1 |
| P4 | 0 | 45 | 88 | Died | Died |
| Week after FVIII plasmid injection | Week after anti-CD3 treatment* | ||||
|---|---|---|---|---|---|
| 1 | 4 | 8 | 2 | 4 | |
| FVIII activities, percentage of normal | |||||
| P1 | 189 | 0 | 0 | 13.3 | 11.1 |
| P2 | 258 | 0 | 0 | 155.4 | 20.5 |
| P3 | 227 | 0 | 0 | 0 | 0 |
| P4 | 304 | 0 | 0 | Died | Died |
| Anti-FVIII inhibitors, BU | |||||
| P1 | 0 | 0.3 | 0.4 | 0 | 0 |
| P2 | 0 | 50 | 62 | 0 | 3 |
| P3 | 0 | 0.3 | 1 | 0 | 0.1 |
| P4 | 0 | 45 | 88 | Died | Died |
Eight weeks after plasmid injection, mice were treated with 40 μg of anti-CD3 for 5 consecutive days. FVIII activities and anti-FVIII inhibitors were monitored before and after anti-CD3 treatment as described in “Gene transfer of FVII into hemophilia A mice with immunomodulation regimen by anti-CD3 antibodies.”
Died indicates that the mouse is deceased.
A 5-day course of anti-CD3 treatment was administered 8 weeks after FVIII plasmid injection treatment.
Discussion
Previously reported data have identified the prominent immune response against FVIII as a major obstacle for successful gene therapy for hemophilia A, whether the FVIII gene was delivered by viral vectors4-7 or naked plasmid DNA.9 Some of the existing strategies, such as using cell type–specific promoters, can only reduce but not eliminate the immune responses.9,30 Here we report a promising strategy to modulate anti-FVIII responses in hemophilia A mice after gene therapy by immunomodulation with anti-CD3 antibody.
Five consecutive injections of modest doses of anti-CD3 delivered concomitantly with plasmid DNA encoding the FVIII gene resulted in long-term, stable FVIII expression in 85% of the treated hemophilia A mice, whereas none developed detectable neutralizing antibodies (inhibitors of FVIII). In comparison, FVIII expression in FVIII plasmid only–treated mice declined to background levels in 2 to 4 weeks and inhibitors of FVIII could be detected as early as 3 to 4 weeks.34,35 Splenic T cells isolated from FVIII plasmid + anti-CD3–treated mice showed no indication of recall proliferation to FVIII stimulation in vitro. Furthermore, with a second plasmid challenge at week 23, when mice had long recovered from the immune suppression of anti-CD3, the treated mice failed to elicit an immune response. Moreover, FVIII challenge under stringent condition (emulsified in CFA) could not break the induced tolerance in FVIII plasmid + anti-CD3 mice. The 2 mice that lost FVIII expression between week 10 and week 15 have had low levels of nonneutralizing anti-FVIII antibodies (supplemental Table 1) that may have led to the eventual loss of FVIII activity. Taken together, these data indicate that anti-CD3 immunomodulation after FVIII plasmid-mediated gene therapy induced partial or complete long-term protection against FVIII-specific immune responses in hemophilia A mice.
As expected, anti-CD3 treatment resulted in depletion of approximately 80% to 90% of CD4+ and CD8+ T cells in spleen and lymph nodes and blood at day 8 after FVIII plasmid injection. This loss of T cells was accompanied by a significant increase in the percentage of CD4+Foxp3+ T cells. However, the absolute number of CD4+Foxp3+ T cells in FVIII plasmid + anti-CD3 mice were comparable with that in FVIII plasmid only mice. The increases in frequency of CD4+Foxp3+ T cells at this early time points could come from 2 sources: (1) anti-CD3 selectively depletes more of the CD4+Foxp3− population within the CD4+ T cells; and (2) anti-CD3 promotes generation of new FVIII-specific CD4+Foxp3+ T cells in the periphery.
The CD4+Foxp3+ T-cell population contains CD25+ (including both CD25hi and CD25low T cells) and CD25− T cells. Interestingly, as shown in Table 1, although the percentage of CD4+CD25+Foxp3+ T cells increased after anti-CD3 treatment, the absolute numbers slightly decreased. This is most probably the result of the selective depletion of activated effector and naive T cells in the CD4 compartment because of higher CD3 expression in these populations (Figure 4B), leading to the enrichment of natural, thymus-originated CD4+CD25+Foxp3+ Tregs. On the other hand, both the percentage and absolute numbers of CD4+CD25−Foxp3+ cells increased significantly in the spleens of FVIII plasmid + anti-CD3–treated mice compared with FVIII plasmid only–treated and naive mice (Table 1). These data indicate that anti-CD3 induced generation of new CD4+CD25−Foxp3+ cells at the early time points after anti-CD3 treatment.
Our data indicated that, at the early time point up to 2 weeks after plasmid injection, the CD4+CD25+Foxp3+ Tregs contain mostly natural, thymus-originated Tregs, which had little contribution to FVIII-specific tolerance induction. This was concluded by 3 separate experiments: (1) CD4+CD25+ T cells isolated from MACS (including CD25hi and CD25low, > 80% Foxp3+) from spleens of FVIII plasmid + anti-CD3–treated mice at week 2 after plasmid injection could not suppress proliferation of FVIII-specific effector T cells (Figure 5A); (2) these CD4+CD25+ T cells could not transfer dominant, FVIII-specific tolerance to syngenic naive mice (data not shown); and (3) depletion of CD4+CD25+ T cells by intravenous injection of anti-CD25 antibodies could not abrogate FVIII-specific tolerance induced by FVIII plasmid + anti-CD3 (Figure 6). Nevertheless, the number of mice in the anti-CD25 group is not large enough to firmly conclude the anti-CD25 antibody does not abrogate anti-CD3 mediated tolerance.
We hypothesize that the newly generated CD4+CD25−Foxp3+ T cells are the Tregs responsible for tolerance induction at the initial stage of FVIII plasmid + anti-CD3 treatment. Studies in mice engineered to express a Foxp3 reporter demonstrated that Foxp3 expression is confined to a subset of αβ T cells and correlates with suppressive activity irrespective of CD25 expression.25-27 In addition, a recent study conducted with SH2 domain-containing inositol 5-phosphatase–deficient mice shows that CD25− T cells express higher Foxp3 levels, and CD4+CD25−Foxp3+ T cells have profound immunosuppressive capacity in vitro and in vivo.37 Moreover, there is now compelling evidence that “adaptive” Tregs may be generated from peripheral CD4+CD25−Foxp3− T cells under well-defined conditions (ie, the type of antigen stimulation, the nature of the APCs, and cytokine milieu).28,32,36,38 Furthermore, we found that the numbers of CD4+CD25+Foxp3+ T cells increased, whereas comparable numbers of CD4+CD25−Foxp3+ T cells decreased in spleens of FVIII plasmid + anti-CD3–treated mice from day 8 to week 4 and 6 after FVIII plasmid injection (Table 1), implying conversion of CD25− cells to CD25+ cells within the CD4+Foxp3+ population. The induced CD4+CD25+Foxp3+ cells are demonstrated to suppress FVIII-specific immune responses at later time points: (1) CD4+CD25+ T cells from spleens of FVIII plasmid + anti-CD3–treated mice at week 6 after plasmid injection suppressed proliferation of FVIII-specific effector T cells (Figure 6B); and (2) the CD4+ T cells transferred dominant, FVIII-specific tolerance to syngenic naive mice (Table 3).
It has been shown that CD3-specific antibody treatment promotes the production of systemic TGF-β1, which is known to induce CD4+Foxp3+ regulatory T cells.28,32,36,38 Belghith et al reported that administration of a neutralizing anti-TGF-β1 antibody abrogated remission of diabetes induced by anti-CD3 antibody,32 indicating the importance of TGF-β1 in maintaining tolerance. In our study, anti-CD3–treated mice exhibited increased TGF-β1 levels in the blood during a period of 8 days after treatment in tolerized mice but not in mice with inhibitors. Altogether, we hypothesize that FVIII plasmid + anti-CD3 treatment induced TGF-β1 dependent conversion of CD4+CD25− cells to “adaptive” CD4+CD25−Foxp3+ Tregs at the periphery at early time point after the treatment; “adaptive” CD4+CD25−Foxp3+ Tregs eventually matured into CD4+CD25+Foxp3+ Tregs to maintain long-term FVIII-specific tolerance. However, we cannot rule out participation of other types of regulatory T cells such as Tr1 in inducing tolerance in our experimental setting.
Waters et al recently reported that anti-CD3 is also able to induce tolerance to FVIII after repeated injections of FVIII proteins in hemophilia A mice via a CD4+CD25+ Treg cell-dependent mechanism.39 Their result is also consistent with our study demonstrating that Tregs are pivotal for establishing FVIII tolerance. However, the development and characteristics of functional antigen-specific Treg cells are different in these 2 studies. We propose that inducible CD4+CD25−/lowFoxp3+ Tregs initiated the tolerance and then matured into CD4+CD25+Foxp3+ Tregs for maintaining long-term tolerance. In their model, CD4+CD25+Foxp3+ Tregs are important for both induction and maintenance of the tolerance. This difference may be the result of the different protocols of anti-CD3 treatment. Waters et al injected anti-CD3 before FVIII protein challenge in which natural CD4+CD25+Foxp3+ Tregs enriched by anti-CD3 treatment may convert to FVIII-specific CD4+CD25+Foxp3+ Tregs on encountering FVIII later; thus, depletion of CD4+CD25+ cells by anti-CD25 abrogated the tolerance induction by anti-CD3 treatment. In our study, we injected anti-CD3 with FVIII plasmid concomitantly; therefore, antigen-specific CD4+CD25−/lowFoxp3+ Tregs were induced at an early time point, and the tolerance was not abrogated by anti-CD25 treatment. In addition, Waters et al used a non-FcR binding, F(ab′)2 form of anti-CD3, and we used a complete form of anti-CD3. Finally, our study demonstrated that, in the majority of the treated mice, FVIII plasmid + anti-CD3 treatment can induce long-term tolerance, which protected the mice from antibody production after the second challenge with either FVIII plasmid or FVIII protein + CFA. Waters et al39 did not perform the second challenge experiment; therefore, it is uncertain whether their protocol of pretreatment with anti-CD3 established the long-term tolerance.
In conclusion, concomitant immunomodulation by anti-CD3 with gene transfer of FVIII plasmid achieved long-term FVIII expression in hemophilia A mice, which was not affected by repeated plasmid DNA applications. FVIII expression greater than 30% of normal human plasma lasted for 34 weeks after gene delivery without detectable anti-FVIII immune responses. Furthermore, anti-CD3 treatment did not affect the host to mount immune responses to T-dependent and T-independent neo-antigens. Anti–human CD3 has been approved by the Food and Drug Administration for clinical use and has been widely applied in transplantation and autoimmune models. The dosage and schedule used in our study are comparable with those used in human trials.14,15,18,23 Anti-CD3 immunomodulation has the potential to be a safe andeffective strategy to prevent FVIII-specific immune responses after gene therapy or protein replacement therapy. Our data also indicate that anti-CD3 can reduce the titers of preexisting inhibitors and will be an excellent candidate in a combination therapy for modulating anti-FVIII immune responses in hemophilia mice with established inhibitors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the National Institutes of Health/National Heart, Lung, and Blood Institute (R01-HL069049 and R01-HL82600) and the Bayer Hemophilia Foundation.
National Institutes of Health
Authorship
Contribution: B.P designed and performed research experiments, analyzed data, and wrote the paper; P.Y. performed experiments; D.J.R. revised the paper; H.D.O. provided helpful ideas and revised the paper; and C.H.M. designed the research project, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carol H. Miao, Department of Pediatrics, Seattle Children's Research Institute and University of Washington, 1900 Ninth Ave, C9S-7, Seattle, WA 98101; e-mail: miao@u.washington.edu.
