

Abstract
The successful reconstitution of adaptive immunity to human cytomegalovirus (CMV) in hematopoietic stem cell transplantation (HSCT) recipients is central to the reduction of viral reactivation-related morbidity and mortality. Here, we characterized the magnitude, specificity, phenotype, function, and clonotypic composition of CMV-specific T-cell responses in 18 donor-recipient pairs both before and after HSCT. The principal findings were: (1) the specificity of CMV-specific T-cell responses in the recipient after HSCT mirrors that in the donor; (2) the maintenance of these targeting patterns reflects the transfer of epitope-specific T-cell clonotypes from donor to recipient; (3) less differentiated CD27+CD57− CMV-specific memory T cells are more likely to persist in the recipient after HSCT compared with more terminally differentiated CD27− CD57+ CMV-specific memory T cells; (4) the presence of greater numbers of less differentiated CD8+ CMV-specific T cells in the donor appears to confer protection against viral reactivation in the recipient after HSCT; and (5) CMV-specific T cells acquire a more differentiated phenotype and a restricted functional profile after HSCT. Overall, these findings define the immunologic factors that influence the successful adoptive transfer of antigen-specific T-cell immunity during HSCT, which enables the identification of recipients at particular risk of CMV reactivation after HSCT.
Introduction
In clinical practice, the adoptive transfer of immunocompetent cells routinely occurs in the setting of allogeneic hematopoietic stem cell transplantation (HSCT) after myeloablative or reduced-intensity conditioning. Thus, the immune system of the recipient after HSCT is donor-derived when successful engraftment and full donor chimerism are achieved. In an effort to exploit the transfer of such immunity for therapeutic purposes, it has been reported that the vaccination of donors before HSCT can augment the reconstitution of antigen-specific responses in the recipient after graft.1-3 Furthermore, in experimental clinical protocols, lymphocytes have been manipulated ex vivo to enrich and/or expand antigen-specific T cells before infusion into the host to confer protective immunity to viruses4-9 and certain tumors.10-12 Along with the adoptive transfer of protective immunity from donor to recipient, however, there is also evidence that autoreactive cells can be transferred as autoimmune or allergic disorders present in the donor have been reported in the recipient after HSCT.13-17
Cytomegalovirus (CMV) reactivation and disease are an important cause of morbidity and mortality in the first year after HSCT; consequently, routine monitoring for reactivations is often used in transplantation protocols with the frequent implementation of preemptive therapy. The measurement of CMV-specific immunity in this setting has generally been limited to a restricted group of antigenic epitopes, for example, the human leukocyte antigen (HLA) A*0201-restricted pp65495-503 peptide, or whole CMV lysate; furthermore, CMV-specific T-cell responses have been identified almost exclusively on the basis of interferon-γ (IFN-γ) secretion with or without coexpression of a surface marker for activation. In general, a more robust reconstitution of CMV-specific T-cell immunity has been noted when both the donor and the recipient are seropositive for CMV.18,19 Several observations indicate that this probably reflects the transfer of effective CMV-specific T cells: (1) the magnitude of CD8+ T-cell responses to selected epitopes, measured physically using peptide-HLA class I tetrameric complexes, has been shown to correlate with a decrease in the frequency of CMV reactivations after HSCT18,20 ; (2) these CMV-specific CD8+ tetramer-binding T cells tend to acquire a more differentiated phenotype with increased secretion of IFN-γ after HSCT21,22 ; and (3) the risk of CMV reactivation is higher when the donor is seronegative and the recipient is seropositive compared with the converse serostatus situation.21,22
In this study, we conducted a comprehensive analysis of CMV-specific T-cell immunity in HLA-matched donors and recipients before and after HSCT to define the key parameters that determine the successful transfer of antiviral T-cell immunity. The total T-cell response to CMV within both the CD4+ and CD8+ subsets was quantified using comprehensive overlapping peptide sets encompassing the 19 immunodominant open-reading frames (ORFs),23 and each individual ORF-specific T-cell response was characterized phenotypically and functionally after antigen stimulation using a polychromatic flow cytometric panel that included 3 phenotypic markers (CD27, CD45RO, and CD57) and the following 5 functional markers: IFN-γ, tumor necrosis factor-α (TNF-α), interleukin-2 (IL-2), macrophage inflammatory protein-1β (MIP-1β), and CD107a, a marker of degranulation.24 Furthermore, we conducted epitope mapping experiments and molecular analyses of T-cell receptor (TCR) usage to define the specificity and clonotypic composition of transferred CMV-specific T-cell populations.
Methods
Patients
All patients provided written permission for their blood to be used for research in accordance with myeloablative stem cell allotransplantation protocols approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute, in accordance with the Declaration of Helsinki. A total of 18 CMV-seropositive patients received radiation-based, myeloablative-matched sibling HSCT depleted of T cells; oral cyclosporine (CsA) was used as graft-versus-host disease (GVHD) prophylaxis. The conditioning regimen in all patients was composed of fludarabine 25 mg/m2 for 5 days, cyclophosphamide 60 mg/kg for 2 days, and total body irradiation at 1200 cGy. A donor lymphocyte infusion (DLI) add-back was scheduled in all patients between days 45 and 100 after HSCT, although this was not administered in 8 of 18 cases. Fluconazole, sulfamethoxazole/trimethoprim, and acyclovir (or derivatives thereof) were used for antimicrobial prophylaxis. Polymerase chain reaction (PCR) chimerism analysis of short tandem repeats was conducted after HSCT in all patients on days 14, 30, 45, 60, 90, and 120, or until donor T cells reach 100%. Reactivation of CMV was assessed on a weekly basis after HSCT with antigenemia assays and/or PCR; CMV antigenemia was measured using the CINA kit (Argene) before March 2005, after which it was replaced by a quantitative PCR for CMV as described previously.25,26 Preemptive treatment was initiated in the event of any detectable CMV antigenemia, a quantifiable quantitative PCR result more than or equal to 1000 genome equivalents/mL, 2 consecutive quantitative PCR results more than 250 genome equivalents/mL, or a low positive result followed by a value in the range of 250 to 1000 genome equivalents/mL. Treatment for CMV reactivation composed induction with ganciclovir or foscarnet for 1 week followed by a maintenance schedule for 1 additional week. Treatment was discontinued when 2 consecutive negative results were achieved; repeated reactivations were treated similarly.
Isolation of PBMCs
Peripheral blood mononuclear cells (PBMCs) were collected from the donors and from the recipients both before and after HSCT; density gradient centrifugation over Ficoll-Paque (MP Biomedicals) was used to prepare the PBMCs, which were subsequently cryopreserved in fetal calf serum (Invitrogen) containing 10% dimethyl sulfoxide (DMSO; Fisher Scientific) according to standard protocols. Immunologic analyses after HSCT were conducted after 100% T-cell donor chimerism was achieved and when sufficient cells were available, usually from a leukapheresis collection. All patients except recipient 6 were aviremic at the time of analysis after HSCT.
Peptides
A total of 138 and 120 peptides that spanned the entire lengths of the pp65 and IE-1 proteins, respectively, were synthesized to more than 70% purity by JPT Peptide Technology; each peptide was 15 amino acids in length and overlapped consecutive peptides by 11 residues. Peptides were dissolved in high-performance liquid chromatography-grade DMSO (Fisher Scientific) at 25 to 100 mg/mL to produce stock solutions. Complete peptide pools for pp65 and IE-1 were prepared that contained individual peptides at equal concentrations. For epitope mapping, each individual peptide was contained in 2 pools and the number of pools was minimum (pp65 and IE-1 matrices).27 For pp65, 24 peptide pools were used; for IE-1, 22 peptide pools were used.27 Consecutive peptides, each 15 amino acids in length with an overlap of 10 residues, were also synthesized for each of 19 CMV ORFs (Mimotopes Pty Ltd) and lyophilized in 96-tube racks; each rack contained 2 controls that were analyzed for quality control purposes by mass spectrometry, high-performance liquid chromatography, and amino acid analysis.23 All peptides were dissolved in DMSO and stored at −20°C. The final concentration of each peptide in all assays was 2 μg/mL.
In vitro cell stimulation
Identical stimulations were performed in the donor and recipient samples. For each experiment, PBMCs were stimulated with complete pp65 and IE-1 pools, 19 ORF peptide pools, and whole HCMV lysate (AD169 strain; Microbix Biosystems Inc). Antigenic peptide stimulation was performed as described previously.28,29 Briefly, purified PBMCs were thawed, resuspended at 1 to 2 × 106 cells/mL in R10 (RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 100 U/mL penicillin G, 100 μg/mL streptomycin sulfate, and 1.7mM sodium glutamine), and rested for at least 2 hours at 37°C in a humidified 5% CO2 atmosphere. Cells were then washed with R10 and readjusted to 1 to 2 × 106 cells/mL. The costimulatory monoclonal antibodies (mAbs) anti-CD28 and anti-CD49d (1 μg/mL final concentration each; BD Biosciences), together with anti-CD107a conjugated to Alexa 680, were added to the cells, which were then transferred in 200-μL aliquots to 96-well round-bottom plates containing each peptide or peptide mix. After incubation for 1 hour at 37°C, brefeldin A (10 μg/mL final concentration; Sigma-Aldrich) and monensin (0.7 μg/mL final concentration; BD Biosciences) were added; the cells were mixed and further incubated for an additional 5 hours. A negative control (costimulatory mAbs alone with no antigen) and a positive control (staphyloccocal enterotoxin B) were included in all experiments. After incubation, the cells were washed in phosphate-buffered saline (PBS) and stained with a violet amine viability dye (Invitrogen). The cells were then washed with PBS containing 1% bovine serum albumin and 0.1% sodium azide, and surface stained with the following directly conjugated mAbs: anti-CD3 APC-Cy7 (BD Biosciences), anti-CD4 quantum dot (QD) 655, anti-CD8 QD585, anti-CD27 PE-Cy5 (Beckman Coulter), anti-CD45RO Texas Red-PE (Beckman Coulter), anti-CD57 QD545, anti-CD14, and anti-CD19 conjugated to Pacific Blue. The cells were then washed again and permeabilized using the cytofix/cytoperm kit (BD Biosciences) before intracellular staining with the following directly conjugated mAbs: anti–IFN-γ FITC, anti–MIP-1β PE, anti–TNF-α PE-Cy7, and anti–IL-2 APC (BD Biosciences). After a final wash, the cells were fixed in PBS containing 1% paraformaldehyde. All QD and Pacific Blue conjugates, together with the anti-CD107a Alexa 680 mAb, were prepared in-house. Unconjugated mAbs were obtained from BD Biosciences; Pacific Blue, Alexa 680, and QDs were obtained from Invitrogen.
Flow cytometric analysis
Cells were analyzed using a modified LSRII flow cytometer (BD Immunocytometry Systems) equipped for the detection of 18 fluorescent parameters. At least 100 000 total events were collected from each sample. Electronic compensation was conducted with antibody capture beads (BD Biosciences) stained separately with each individual mAb. Data analysis was performed using FlowJo software, Version 8.6 (TreeStar). Initial gating of each sample used a forward scatter area versus a side scatter height plot to isolate small lymphocytes followed by a forward scatter area versus a forward scatter height plot to gate out cell aggregates. To minimize background levels of staining caused by nonspecific binding events, CD14+ cells, CD19+ cells, and dead cells were excluded using a dump channel. Non-T cells were excluded by gating on CD3+ cells and naive cells were excluded by gating out CD27+CD45RO− cells. For subset analyses, CD4+ T cells were selected by first gating out CD8+ cells and subsequently gating on CD4+ cells; conversely, CD8+ T cells were selected by first gating out CD4+ cells and subsequently gating on CD8+ cells. All T-cell markers were plotted versus IFN-γ to observe and include stimulated cells that had down-regulated surface marker expression (supplemental Figure 1, available on the Blood website; see the Supplemental Materials link at the top of the online article). For the purposes of this study, we classified cells as naive (CD27+CD45RO−), CD27+ memory (CD27+CD45RO+CD57−), CD27− memory (CD27−), or CD57+ memory (CD27−CD57+).30 Boolean combinations of single function gates were created using FlowJo software to determine the total response to CMV and the frequency of each response based on all possible combinations of cytokine and CD107a expression. Background responses detected in negative control tubes were subtracted from those detected in stimulated samples for every specific functional combination. Because of differences in the frequency of the memory T-cell population in the donor and recipient after HSCT, the frequency of CMV-specific T cells is shown as the percentage of CD4+ and CD8+ memory T cells. For representational analyses, negative values were set to zero.
Cell sorting
All sorts were performed on stained cells fixed with 1% paraformaldehyde using a modified FACSAria flow cytometer (BD Immunocytometry Systems). PBMCs were prepared and rested as described in “In vitro cell stimulation,” and then incubated overnight in R10 supplemented with anti-CD28 and anti-CD49d mAbs (1 μg/mL final concentration each; BD Biosciences), brefeldin A (10 μg/mL; Sigma-Aldrich), and individual pp65 or IE-1 peptides at 2 μg/mL. CD8+ T cells expressing IFN-γ after overnight peptide stimulation were sorted into dry collection tubes and the cell pellets were frozen at −80°C (supplemental Figure 2). Similar sorts were conducted for both CD4+ and CD8+ T cells after stimulation with CMV ORF peptide pools, as described in “Flow cytometric analysis.” Instrument setup in all cases was performed according to the manufacturer's instructions.
Clonotype analysis
Functional T cells sorted from antigen-stimulated cultures were analyzed for TCR β-chain (TCRB) gene rearrangements with a genomic DNA-based clonotyping assay as described previously in detail.31,32 Briefly, rearranged TCRB fragments were amplified using a hemi-nested touchdown PCR in 3 different tubes. PCR products of 250 to 300 bp were excised, purified, ligated, and transformed as described elsewhere.33,34 Selected colonies were amplified by PCR using standard M13 primers and then sequenced from an insert-specific primer using fluorescent dye terminator chemistry (Applied Biosystems). All sequences that were only found once in each cell population were disregarded. Data analysis was performed using Sequencher, Version 4.2 (Gene Codes Corporation). The International Immunogenetics nomenclature system is used throughout.35
Statistical analysis
The 2-tailed Mann-Whitney U test was used to compare the medians between groups, and the Spearman rank correlation coefficient was used to measure correlations. Comparisons were performed using GraphPad Prism, Version 5.00, and considered significant when P was less than .05.
Results
Patient details
A detailed analysis of CMV-specific T-cell immunity was conducted in 18 CMV-seropositive donor-recipient pairs. The donor cells were analyzed before HSCT, and the recipient cells were analyzed before and after HSCT, the latter once 100% donor T-cell chimerism had been achieved. Engraftment was universal, and all patients converted to full donor chimerism. The median age of the cohort was 31 years, and the median CD34+ cell dose was 5.34 × 106/kg (Table 1). The graft was T cell–depleted in all patients; a fixed dose of 2 × 104 T cells/kg was infused along with the CD34+ cells. A DLI was administered in 10 cases between 43 and 96 days after HSCT; 8 patients with GVHD or CsA intolerance did not receive a DLI (Table 1). A positive CMV antigenemia or PCR positivity of greater than 250 genome equivalents/mL was detected in 12 patients after HSCT (Table 1). The median time to initial CMV reactivation was 30 days (range, 17-68 days) after HSCT. In all but 1 patient, CMV reactivation occurred before DLI. Of the 7 patients with grade II-IV GVHD, 2 did not reactivate CMV, 2 had initial CMV reactivation before the onset of GVHD, and 3 experienced CMV reactivation after the onset of GVHD. A 100% donor T-cell chimerism was achieved at a median of 45 days (range, 29-118 days), and immunologic analysis was performed at a median of 119 (range, 61-171) days after HSCT, when most were outpatients on a CsA taper regimen and low-dose corticosteroids (Table 1). The median absolute lymphocyte counts were 2171 cells/μL (range, 1449-4011 cells/μL) in the donor, 1117 cells/μL (range, 338-5628 cells/μL) in the recipient before HSCT, and 1205 cells/μL (299-2954 cells/μL) in the recipient after HSCT. The corresponding absolute CD4 counts were 1015 cells/μL (range, 523-1710 cells/μL), 454 cells/μL (range, 75-2644 cells/μL), and 213 cells/μL (range, 69-1055 cells/μL), respectively; the corresponding absolute CD8 counts were 410 cells/μL (range, 228-790 cells/μL), 234 cells/μL (range, 33-1175 cells/μL), and 282 (36-1155 cells/μL), respectively.
Patient and transplantation characteristics
| ID | Diagnosis | Age, y | CD34+ dose, 106/kg | T-cell dose, 104/kg | Conditioning | GVHD prophylaxis | DLI (day) | Acute GVHD, grade | Day of 100% donor T-cell chimerism | Day after HSCT of CMV analysis | No. of CMV reactivations | Days of CMV positivity | IST on day of CMV analysis after HSCT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CML | 41 | 4.22 | 2 | Flu/Cy/TBI | CsA | 63 | — | 90 | 120 | 0 | 0 | CsA 125 mg bid; Pred 10 mg qod |
| 2 | CML | 29 | 4.84 | 2 | Flu/Cy/TBI | CsA | — | — | 34 | 171 | 1 | 7 | CsA 25 mg bid; Pred 5 mg qd |
| 3 | AML | 32 | 4.36 | 2 | Flu/Cy/TBI | CsA | 45 | — | 90 | 120 | 2 | 21 | Pred 10 mg qd |
| 4 | CML | 21 | 4.86 | 2 | Flu/Cy/TBI | CsA | — | S (II) | 45 | 62 | 0 | 0 | CsA 50 mg bid; Pred 15 mg qod |
| 5 | ALL | 18 | 5.3 | 2 | Flu/Cy/TBI | CsA | — | G (IV) | 34 | 61 | 1 | 7 | Sirolimus 2.3 mg qd; Methylpred 60 mg qd |
| 6 | CML | 31 | 10 | 2 | Flu/Cy/TBI | CsA | 44 | — | 62 | 118 | 3 | 28 | CsA 75 mg bid |
| 7 | AML | 40 | 6.16 | 2 | Flu/Cy/TBI | CsA | 96 | — | 45 | 126 | 3 | 28 | CsA 100 mg bid |
| 8 | AML | 31 | 7.28 | 2 | Flu/Cy/TBI | CsA | 63 | — | 11 | 120 | 9 | 119 | CsA 100 mg bid |
| 9 | MF | 33 | 9.36 | 2 | Flu/Cy/TBI | CsA | 70 | — | 44 | 118 | 1 | 21 | CsA 50 mg bid; hydrocortisone 15 mg qd |
| 10 | ALL | 17 | 7.22 | 2 | Flu/Cy/TBI | CsA | 46 | — | 46 | 122 | 0 | 0 | CsA 75 mg bid: Pred 10 mg qod |
| 11 | AML | 30 | 5.17 | 2 | Flu/Cy/TBI | CsA | 43 | — | 43 | 84 | 0 | 0 | CsA 75 mg bid; Pred 20 mg qod |
| 12 | AML | 29 | 8.28 | 2 | Flu/Cy/TBI | CsA | — | S/L (II) | 29 | 120 | 3 | 28 | CsA 125 mg bid |
| 13 | CML | 27 | 3.91 | 2 | Flu/Cy/TBI | CsA | 63 | — | 62 | 155 | 0 | 0 | CsA 50 mg bid; Pred 10 mg qod |
| 14 | MDS | 39 | 4.27 | 2 | Flu/Cy/TBI | CsA | 55 | — | 116 | 133 | 1 | 16 | CsA 75 mg bid; Pred 20 mg qd |
| 15 | AML | 27 | 5.38 | 2 | Flu/Cy/TBI | CsA | — | S/G (II) | 36 | 63 | 1 | 7 | CsA 100 mg bid; Pred 5 mg qod |
| 16 | ALL | 32 | 5.80 | 2 | Flu/Cy/TBI | CsA | — | L/G (II) | 45 | 62 | 3 | 21 | Tacro 0.5 mg qd; Pred 5 mg qd |
| 17 | ALL | 36 | 3.36 | 2 | Flu/Cy/TBI | CsA | — | S (II) | 60 | 71 | 2 | 19 | CsA 125 mg bid |
| 18 | AML | 39 | 9.10 | 2 | Flu/Cy/TBI | CsA | — | S (II) | 30 | 82 | 0 | 0 | CsA 25 mg qd; Pred 5 mg qd |
| ID | Diagnosis | Age, y | CD34+ dose, 106/kg | T-cell dose, 104/kg | Conditioning | GVHD prophylaxis | DLI (day) | Acute GVHD, grade | Day of 100% donor T-cell chimerism | Day after HSCT of CMV analysis | No. of CMV reactivations | Days of CMV positivity | IST on day of CMV analysis after HSCT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CML | 41 | 4.22 | 2 | Flu/Cy/TBI | CsA | 63 | — | 90 | 120 | 0 | 0 | CsA 125 mg bid; Pred 10 mg qod |
| 2 | CML | 29 | 4.84 | 2 | Flu/Cy/TBI | CsA | — | — | 34 | 171 | 1 | 7 | CsA 25 mg bid; Pred 5 mg qd |
| 3 | AML | 32 | 4.36 | 2 | Flu/Cy/TBI | CsA | 45 | — | 90 | 120 | 2 | 21 | Pred 10 mg qd |
| 4 | CML | 21 | 4.86 | 2 | Flu/Cy/TBI | CsA | — | S (II) | 45 | 62 | 0 | 0 | CsA 50 mg bid; Pred 15 mg qod |
| 5 | ALL | 18 | 5.3 | 2 | Flu/Cy/TBI | CsA | — | G (IV) | 34 | 61 | 1 | 7 | Sirolimus 2.3 mg qd; Methylpred 60 mg qd |
| 6 | CML | 31 | 10 | 2 | Flu/Cy/TBI | CsA | 44 | — | 62 | 118 | 3 | 28 | CsA 75 mg bid |
| 7 | AML | 40 | 6.16 | 2 | Flu/Cy/TBI | CsA | 96 | — | 45 | 126 | 3 | 28 | CsA 100 mg bid |
| 8 | AML | 31 | 7.28 | 2 | Flu/Cy/TBI | CsA | 63 | — | 11 | 120 | 9 | 119 | CsA 100 mg bid |
| 9 | MF | 33 | 9.36 | 2 | Flu/Cy/TBI | CsA | 70 | — | 44 | 118 | 1 | 21 | CsA 50 mg bid; hydrocortisone 15 mg qd |
| 10 | ALL | 17 | 7.22 | 2 | Flu/Cy/TBI | CsA | 46 | — | 46 | 122 | 0 | 0 | CsA 75 mg bid: Pred 10 mg qod |
| 11 | AML | 30 | 5.17 | 2 | Flu/Cy/TBI | CsA | 43 | — | 43 | 84 | 0 | 0 | CsA 75 mg bid; Pred 20 mg qod |
| 12 | AML | 29 | 8.28 | 2 | Flu/Cy/TBI | CsA | — | S/L (II) | 29 | 120 | 3 | 28 | CsA 125 mg bid |
| 13 | CML | 27 | 3.91 | 2 | Flu/Cy/TBI | CsA | 63 | — | 62 | 155 | 0 | 0 | CsA 50 mg bid; Pred 10 mg qod |
| 14 | MDS | 39 | 4.27 | 2 | Flu/Cy/TBI | CsA | 55 | — | 116 | 133 | 1 | 16 | CsA 75 mg bid; Pred 20 mg qd |
| 15 | AML | 27 | 5.38 | 2 | Flu/Cy/TBI | CsA | — | S/G (II) | 36 | 63 | 1 | 7 | CsA 100 mg bid; Pred 5 mg qod |
| 16 | ALL | 32 | 5.80 | 2 | Flu/Cy/TBI | CsA | — | L/G (II) | 45 | 62 | 3 | 21 | Tacro 0.5 mg qd; Pred 5 mg qd |
| 17 | ALL | 36 | 3.36 | 2 | Flu/Cy/TBI | CsA | — | S (II) | 60 | 71 | 2 | 19 | CsA 125 mg bid |
| 18 | AML | 39 | 9.10 | 2 | Flu/Cy/TBI | CsA | — | S (II) | 30 | 82 | 0 | 0 | CsA 25 mg qd; Pred 5 mg qd |
The DLI dose was 107/kg. Grade II to IV GVHD is shown. Some patients who did not experience grade II to IV GVHD were on low-dose corticosteroids at the time of immunologic analysis after HSCT for adrenal insufficiency (1 patient), mild skin GVHD (5 patients), or oral GVHD after day 100 (2 patients).
CML indicates chronic myelogenous leukemia; AML, acute myelogenous leukemia; ALL, acute lymphocytic leukemia; MDS, myelodysplasia; MF, myelofibrosis; Flu, fludarabine 25 mg/m2 for 5 days; Cy, cyclophosphamide 60 mg/kg for 2 days; TBI, total body irradiation at 1200 cGy; GVHD, graft-versus-host disease; DLI, donor lymphocyte infusion; CMV, cytomegalovirus; HSCT, hematopoietic stem cell transplantation; S, skin; G, gut; L, liver; IST, immunosuppressive therapy; CsA, cyclosporine; Tacro, tacrolimus; Pred, prednisone; Methylpred, methylprednisolone; qd, once a day; bid, twice a day; qod, every other day; and —, not applicable.
Magnitude of CMV-specific T-cell responses in donors and recipients
To approximate the total frequency of CMV-specific T cells, ORF peptide mixes that account for the majority of the CMV epitopes targeted by specific CD4+ and CD8+ memory T cells were used to stimulate PBMCs from the donors and recipients.23 To ensure that individual cells were counted as single events regardless of their functional profile, all possible 31 Boolean combinations of cytokines and CD107a expression (where at least a single response was observed) were added for each ORF and the total frequency of CMV-specific T cells was determined by the summation of responses to the 19 ORFs for both CD4+ and CD8+ subsets; the flow cytometric gating strategy for these functional analyses is shown in supplemental Figure 1. In most cases, the total frequency of CD4+ and CD8+ CMV-specific T cells in the recipient after HSCT was higher compared with the donor (Figure 1). This increase in frequency corresponded to an increase in the absolute numbers of CMV-specific T cells in most recipients after HSCT (supplemental Figures 3-5). In 4 donor-recipient pairs (ID nos. 4, 8, 11, and 14), a decrease in the frequency of the CD4+ T-cell response after HSCT was observed compared with the donor; for CD8+ T-cell responses, a similar decrease was observed in 2 donor-recipient pairs (ID nos. 8 and 9; Figure 1). The median frequency of CMV-specific CD4+ T cells was 2.60% (range, 0.47%-6.92%) in the donor and 5.15% (range, 0.11%-22.65%) in the recipient after HSCT; for CD8+ T cells, the median frequency was 7.74% (range, 0.56%-18.24%) in the donor and 15.82% (range, 2.64%-70%) in the recipient after HSCT. The median absolute numbers of CMV-specific CD4+ T cells were 24 cells/μL (range, 4-48 cells/μL) in the donor and 18 cells/μL (range, 0-99 cells/μL) in the recipient after HSCT; the corresponding median absolute numbers of CD8+ T cells were 25 cells/μL (range, 3-97 cells/μL) in the donor and 47 cells/μL (range, 2-321 cells/μL) in the recipient after HSCT. Thus, the frequency of donor-derived CMV-specific T cells increased in the recipients after HSCT in the majority of cases.

Total CMV-specific CD4+ and CD8+ T-cell responses in donors, recipients before HSCT, and recipients after HSCT. CD4+ (top panel) and CD8+ (bottom panel) T-cell responses are shown in donors (green), recipients before HSCT (red), and recipients after HSCT (yellow). After stimulation for 6 hours, the frequency of CMV-specific memory T cells was calculated using all possible Boolean combinations (where at least a single response was present) of cytokines (MIP-1β, IL-2, TNF-α, and IFN-γ) and CD107a expression for each ORF, and the total frequency was determined by the summation of responses to the 19 ORFs for both CD4+ and CD8+ subsets (after background subtraction). *Patients who received a DLI.
Total CMV-specific CD4+ and CD8+ T-cell responses in donors, recipients before HSCT, and recipients after HSCT. CD4+ (top panel) and CD8+ (bottom panel) T-cell responses are shown in donors (green), recipients before HSCT (red), and recipients after HSCT (yellow). After stimulation for 6 hours, the frequency of CMV-specific memory T cells was calculated using all possible Boolean combinations (where at least a single response was present) of cytokines (MIP-1β, IL-2, TNF-α, and IFN-γ) and CD107a expression for each ORF, and the total frequency was determined by the summation of responses to the 19 ORFs for both CD4+ and CD8+ subsets (after background subtraction). *Patients who received a DLI.
Targeting patterns of CMV-specific T-cell responses in donors and recipients
Overall, the CMV-specific CD4+ and CD8+ T-cell responses tended to target the same ORFs in the donor and the recipient after HSCT (Figure 2A; supplemental Figures 3-5). In some cases, the CMV-specific T-cell response targeted relatively few ORFs; in others, the response was more broadly directed against multiple ORFs (Figure 2A; supplemental Figures 3-5). Of note, the ORF-specific T-cell responses detected in some recipients after HSCT were not always present in the donor, thereby suggesting that the donor response could lie below the limit of detection by flow cytometry or that de novo CMV-specific T-cell clonotypes were generated in the recipient after HSCT.36

Contribution of ORF-specific T-cell response to the total CMV-specific T-cell response in the donor and recipient after HSCT. (A) Targeting patterns of CMV-specific memory CD4+ (left panels) and CD8+ (right panels) T-cell responses in representative donor-recipient pairs. The summation of responses measured by 5 functional outputs is shown for each ORF (CD4+ and CD8+) tested and for whole CMV lysate (CD4+ only); donor (green), recipient before HSCT (red), and recipient after HSCT (yellow). Response breadth (ie, the number of targeted ORFs) and dominance patterns (ie, the ORF-specific responses representing the majority of the total response) showed substantial variability. In some cases, pp65 and IE-1 were preferentially targeted (eg, ID 1 and ID 3 CD8+ T-cell responses); in other cases, the predominant responses were directed against different ORFs (eg, ID 7 CD8+ T-cell responses). In some cases, the CD4+ T-cell responses to whole CMV lysate were less than the summation of the ORFs, which may reflect the negative effect of freeze/thawing in lysate processing for peptide recognition. *Patients who received a DLI. (B) Contribution of each ORF-specific response to the total CD4+ (left panel) and CD8+ (right panel) CMV-specific T-cell response plotted for donors against recipients after HSCT. In each case, the ORF-specific response was normalized such that the contribution of each targeted ORF (in percentage) to the total CMV-specific T-cell response was calculated. Only the normalized data are shown.
Contribution of ORF-specific T-cell response to the total CMV-specific T-cell response in the donor and recipient after HSCT. (A) Targeting patterns of CMV-specific memory CD4+ (left panels) and CD8+ (right panels) T-cell responses in representative donor-recipient pairs. The summation of responses measured by 5 functional outputs is shown for each ORF (CD4+ and CD8+) tested and for whole CMV lysate (CD4+ only); donor (green), recipient before HSCT (red), and recipient after HSCT (yellow). Response breadth (ie, the number of targeted ORFs) and dominance patterns (ie, the ORF-specific responses representing the majority of the total response) showed substantial variability. In some cases, pp65 and IE-1 were preferentially targeted (eg, ID 1 and ID 3 CD8+ T-cell responses); in other cases, the predominant responses were directed against different ORFs (eg, ID 7 CD8+ T-cell responses). In some cases, the CD4+ T-cell responses to whole CMV lysate were less than the summation of the ORFs, which may reflect the negative effect of freeze/thawing in lysate processing for peptide recognition. *Patients who received a DLI. (B) Contribution of each ORF-specific response to the total CD4+ (left panel) and CD8+ (right panel) CMV-specific T-cell response plotted for donors against recipients after HSCT. In each case, the ORF-specific response was normalized such that the contribution of each targeted ORF (in percentage) to the total CMV-specific T-cell response was calculated. Only the normalized data are shown.
The majority of previous studies have primarily focused on responses to pp65 and IE-1. Indeed, in some persons, responses to UL83 (pp65) and UL123 (IE-1) contributed significantly to the total CMV-specific T-cell response (Figure 2A; supplemental Figures 3-5). However, responses to these ORFs were minimal or absent in other donor-recipient pairs, in which other ORFs were preferentially targeted. As a frequency of the total CD4+ T-cell response to CMV, the combined responses to UL83 (pp65) and UL123 (IE-1) amounted to a median of 15% (range, 0.1%-38.5%) in the donor, 14.2% (range, 0%-71.2%) in the recipient before HSCT, and 17.5% (range, 0.64%-46.7%) in the recipient after HSCT; the corresponding numbers for the CD8+ T-cell response to CMV were 25.4% (range, 4.5%-71.3%) in the donor, 19.3% (range, 0%-65.7%) in the recipient before HSCT, and 16.9% (range, 0%-62.5%) in the recipient after HSCT. Thus, the measurement of responses to pp65 and IE-1 alone can greatly underestimate the frequency of the total CMV-specific T-cell response.
To compare the contribution of each targeted ORF to the total CMV-specific T-cell response in donors and recipients, the frequency of each ORF-specific response was normalized such that the percentage of the total response represented by each targeted ORF was calculated and the summation of these relative contributions totaled 100% of the overall CMV-specific T-cell response. Among the ORF-specific T-cell responses that were greater than 0 in the recipient after HSCT relative to the respective donor, a correlation between the contribution of each targeted ORF to the total response in the donor and in the recipient after HSCT was observed for both CD4+ (r = 0.47, P < .001; Figure 2B) and CD8+ (r = 0.55, P < .001; Figure 2B) CMV-specific T-cell responses. Interestingly, similar correlations were observed between donors and recipients before HSCT (all were matched at the HLA-A, B, and DR loci) for both CD4+ (r = 0.45, P < .001) and CD8+ (r = 0.52, P < .001) CMV-specific T-cell responses (data not shown). Thus, after HSCT, the breadth and specificity of the reconstituted CMV-specific T-cell response in the recipient recapitulate that of the donor.
Clonotypic composition of CMV-specific T-cell responses in donors and recipients
To determine the clonotypic composition of individual ORF-specific responses, CD4+ and CD8+ T cells that produced IFN-γ in ORF peptide pool stimulations were sorted from 5 donor-recipient pairs after overnight stimulation; TCRB gene rearrangements within the sorted functional populations were analyzed using a multiplex DNA-based molecular approach (Table 2).31,32 In general, the clonotypic repertoire for individual ORF-specific T-cell populations was oligoclonal; this is consistent with previous studies of both CD4+ and CD8+ CMV-specific T-cell populations.37,38 In all donor-recipient pairs, clonotypes that were identified in the donor were also present in the recipient after HSCT for both CD4+ and CD8+ ORF-specific T-cell populations (Table 2).
Clonal composition of CMV-specific T cells to different ORFs, pp65, and IE-1 in donor-recipient pairs
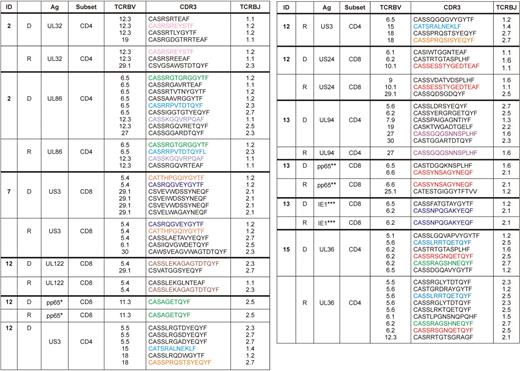
D indicates donor; and R, recipient after HSCT.
For pp65 and IE-1 stimulations (supplemental Figure 2), the dominant peptide targeted in matrix experiments was used as the stimulatory antigen in each case (Table 3): *EDVPSGKLFMHVTLG, **ARNLVPMVATVQGQN, ***LSEFCRVLCCYVLEE. For other data shown, complete ORF peptide pools were used as the stimulus.
Color codes denote identical clonotypes between each donor-recipient pair.
To confirm the transfer of donor-derived CMV-specific T cells to the recipient during HSCT, epitope mapping experiments were performed in selected cases for CD8+ T-cell populations more than 0.2% in the donor and recipient that produced IFN-γ in response to pp65 and/or IE-1 peptide pools. Of the 4 donor-recipient pairs tested, matrix analyses demonstrated that the same dominant peptide was targeted in the donor and in the recipient after HSCT, thereby suggesting the transfer of epitope-specific CD8+ T-cell clonotypes from donor to recipient (Table 3). Furthermore, immunodominance hierarchies were also maintained from donor to recipient with respect to the subdominant intraprotein epitope-specific responses. Characterization of TCRB gene rearrangement profiles within the dominant epitope-specific CD8+ T-cell responses, conducted as described for the ORF-specific T-cell responses, again revealed the presence of the same clonotypes in the corresponding donor-recipient pairs (Table 2). Thus, the reconstitution of CMV-specific T-cell immunity in the recipient after HSCT is based on the transfer of donor-derived T-cell clonotypes.
Epitope mapping of the dominant CD8+ CMV-specific responses to pp65 and IE-1 in the donor and recipient after HSCT
| Donor-recipient after HSCT pair | Antigen | 15-mer |
|---|---|---|
| 3 | pp65 | SAAERKHRHLPVADA |
| 10 | pp65 | ARNLVPMVATVQGQN |
| 12 | pp65 | EDVPSGKLFMHVTLG |
| 13 | pp65 | ARNLVPMVATVQGQN |
| 3 | IE-1 | KVFAQYILGADPLRV |
| 13 | IE-1 | LSEFCRVLCCYVLEE |
| Donor-recipient after HSCT pair | Antigen | 15-mer |
|---|---|---|
| 3 | pp65 | SAAERKHRHLPVADA |
| 10 | pp65 | ARNLVPMVATVQGQN |
| 12 | pp65 | EDVPSGKLFMHVTLG |
| 13 | pp65 | ARNLVPMVATVQGQN |
| 3 | IE-1 | KVFAQYILGADPLRV |
| 13 | IE-1 | LSEFCRVLCCYVLEE |
Phenotypic characterization of CMV-specific T-cell responses in donors and recipients
In our initial analysis, we observed that the frequency of some ORF-specific T-cell responses in the donor increased in the recipient after HSCT, whereas others decreased. Further characterization of these responses was enabled by the inclusion of phenotypic markers (CD27, CD45RO, and CD57) in the polychromatic flow cytometric panel used for functional screening (supplemental Figure 6). To minimize any background bias, phenotypic characterization was only performed for CMV-specific T cells that produced IFN-γ, TNF-α, or IL-2; this analysis was conducted for responses in the donor with magnitudes more than 0.2% that either increased or decreased more than 2-fold in the recipient after HSCT (responses in the recipient < 0.05% were not included). A preferential phenotype was not observed for any of the ORF-specific T-cell populations. A significant positive correlation was observed between the frequency of CMV-specific CD8+CD27+ memory T cells in the donor and the likelihood of the same ORF-specific response increasing in the recipient after HSCT (r = 0.63, P < .001; Figure 3A); significant negative correlations were observed between the frequencies of CMV-specific CD8+CD27− (r = − 0.67, P < .001; Figure 3A) and CD8+CD57+ (r = − 0.45, P = .001; Figure 3A) memory T cells in the donor and the likelihood of the same ORF-specific response decreasing in the recipient after HSCT. These correlations were not as strong for the ORF-specific CD4+ memory T-cell responses. Thus, CMV-specific T-cell populations that exhibited less differentiated phenotypes in the donor were more likely to persist and increase after transfer to the recipient.

Phenotypic analysis of donor CMV ORF-specific memory T-cell responses in relation to CMV reactivations and magnitude of the corresponding responses in the recipient after HSCT. (A) For donor ORF-specific T-cell populations that were less differentiated (CD27+CD45RO+CD57−), there was a greater probability of persistence and expansion (increased fold change) in the recipient after HSCT. For donor ORF-specific T-cell populations that were more differentiated (CD27− memory; CD27−CD57+ memory), there was a greater probability of contraction (decreased fold change) in the recipient after HSCT. (B) In donors with a higher frequency of CD8+ CMV-specific CD27+ memory T-cell responses, fewer CMV reactivations were observed in the recipient after HSCT. Conversely, in donors with a higher frequency of more differentiated CD8+ CMV-specific memory T-cell responses (CD27−), a greater number of CMV reactivations were observed in the recipient after HSCT. No correlations between the phenotype of donor CMV-specific CD4+ T cells and the incidence of CMV reactivation in the recipient after HSCT were observed. The patients who reactivated CMV are shown in Table 1.
Phenotypic analysis of donor CMV ORF-specific memory T-cell responses in relation to CMV reactivations and magnitude of the corresponding responses in the recipient after HSCT. (A) For donor ORF-specific T-cell populations that were less differentiated (CD27+CD45RO+CD57−), there was a greater probability of persistence and expansion (increased fold change) in the recipient after HSCT. For donor ORF-specific T-cell populations that were more differentiated (CD27− memory; CD27−CD57+ memory), there was a greater probability of contraction (decreased fold change) in the recipient after HSCT. (B) In donors with a higher frequency of CD8+ CMV-specific CD27+ memory T-cell responses, fewer CMV reactivations were observed in the recipient after HSCT. Conversely, in donors with a higher frequency of more differentiated CD8+ CMV-specific memory T-cell responses (CD27−), a greater number of CMV reactivations were observed in the recipient after HSCT. No correlations between the phenotype of donor CMV-specific CD4+ T cells and the incidence of CMV reactivation in the recipient after HSCT were observed. The patients who reactivated CMV are shown in Table 1.
The total frequency of CD27+ and CD57+ CMV-specific memory T cells in the donors was further analyzed in relation to the incidence of CMV reactivation in the recipients after HSCT. In patients who experienced no CMV reactivation after HSCT, the median frequencies of CMV-specific T cells in the donors were 2.32% (range, 0.42%-3.2%) for CD4+ memory responses and 6.67% (range, 0.56%-18.2%) for CD8+ memory responses; in patients who experienced one or more reactivation, the corresponding median frequencies in the donor were 2.85% (range, 0.53%-6.92%) and 7.74% (range, 1.75%-16.7%), respectively. These observations suggest that the total frequency of CMV-specific memory T cells in the donor was not associated with protection in the recipient after HSCT. There were no ORFs that were preferentially targeted in the donors of recipients who did not experience CMV reactivation after HSCT. The frequency of CD4+CD27+ or CD4+CD57+ CMV-specific memory T cells did not appear to correlate with the incidence of viral reactivation in the recipient after HSCT; however, a higher frequency of CD8+CD27+ and a lower frequency of CD8+CD27− CMV-specific memory T cells were observed in donors of recipients who did not experience CMV reactivation compared with donors of recipients who experienced one or more reactivation (P < .05; Figure 3B). In addition, a higher frequency of IL-2–producing CMV-specific CD8+ T cells was observed in donors of recipients who did not experience CMV reactivation after HSCT in similar analyses (P = .01, data not shown). Furthermore, CD8+ CMV-specific T cells were more differentiated (CD27−) in donors of recipients who experienced one or more reactivation after HSCT (Figure 3B). The lowest frequencies of CMV-specific CD27+ memory T cells were observed in donor ID 8; both CD4+ and CD8+ CMV-specific T-cell responses decreased after HSCT, and the corresponding recipient experienced the highest number of CMV reactivations in our cohort (Table 1).
In additional analyses, donor ORF-specific T-cell responses were compared phenotypically with the corresponding responses in recipients after HSCT (Figure 4A). For both CD4+ and CD8+ T cells, the ORF-specific responses in recipients after HSCT were more differentiated compared with the corresponding responses in the respective donors (Figure 4A). Thus, transferred CMV-specific T-cell populations with less differentiated phenotypes appear to confer a reduced likelihood of CMV reactivation in recipients after HSCT; furthermore, a significant degree of differentiation occurred in all CMV-specific T-cell populations after HSCT.

Changes in phenotype and cytokine production of CMV ORF-specific T cells in the donor and recipient after HSCT. (A) Phenotype of CMV ORF-specific responses in donors and recipients after HSCT. Matched ORF-specific responses to CMV acquired a more differentiated phenotype after HSCT, with significant increases in both CD27− memory and CD57+ memory T-cell populations in both the CD4+ (left panels) and CD8+ (right panels) subsets; similarly, significant decreases were observed in the corresponding CD27+ memory T-cell populations. (B) Analysis of the contribution of each cytokine and CD107a to the increment in CMV-specific T-cell responses after HSCT is shown in the upper panels. Each of the 5 independent functions was analyzed for each ORF-specific T-cell response after HSCT and compared with the corresponding response in the donor. For CD4+ T-cell responses, IFN-γ secretion increased in the recipient after HSCT (P = .024); for CD8+ T-cell responses, an increment in CD107a mobilization and the production of IFN-γ, MIP-1β, and TNF-α was observed in the recipient after HSCT. When T-cell responses were categorized on the basis of the number of functions elicited on CMV antigen encounter (bottom panels), bifunctional responses increased within both the CD4+ and CD8+ subsets after HSCT (P = .017 and P = .003, respectively). The median is depicted in red. Only frequencies ≥ 0.01 are shown for graphical representation. D indicates donor; and R, recipient after HSCT. *P < .05, **P < .01 (Mann-Whitney U test).
Changes in phenotype and cytokine production of CMV ORF-specific T cells in the donor and recipient after HSCT. (A) Phenotype of CMV ORF-specific responses in donors and recipients after HSCT. Matched ORF-specific responses to CMV acquired a more differentiated phenotype after HSCT, with significant increases in both CD27− memory and CD57+ memory T-cell populations in both the CD4+ (left panels) and CD8+ (right panels) subsets; similarly, significant decreases were observed in the corresponding CD27+ memory T-cell populations. (B) Analysis of the contribution of each cytokine and CD107a to the increment in CMV-specific T-cell responses after HSCT is shown in the upper panels. Each of the 5 independent functions was analyzed for each ORF-specific T-cell response after HSCT and compared with the corresponding response in the donor. For CD4+ T-cell responses, IFN-γ secretion increased in the recipient after HSCT (P = .024); for CD8+ T-cell responses, an increment in CD107a mobilization and the production of IFN-γ, MIP-1β, and TNF-α was observed in the recipient after HSCT. When T-cell responses were categorized on the basis of the number of functions elicited on CMV antigen encounter (bottom panels), bifunctional responses increased within both the CD4+ and CD8+ subsets after HSCT (P = .017 and P = .003, respectively). The median is depicted in red. Only frequencies ≥ 0.01 are shown for graphical representation. D indicates donor; and R, recipient after HSCT. *P < .05, **P < .01 (Mann-Whitney U test).
Functionality of CMV-specific T-cell responses in donors and recipients
To determine the impact of HSCT on the functionality of CMV-specific T-cell responses, the frequency of each cytokine and CD107a for each ORF-specific response in recipients after HSCT was analyzed independently and compared with the same ORF-specific response in the corresponding donors (Figure 4B). For CD4+ T cells, the secretion of IFN-γ (P = .009) and TNF-α (P = .046) increased after HSCT; increments in other functional outputs did not achieve statistical significance. For CD8+ T cells, significant increases in the secretion of IFN-γ (P = .029), MIP-1β (P = .001), TNF-α (P = .045), and the expression of CD107a (P = .045) were observed in the recipient after HSCT (Figure 4B top panels). There were also substantial changes in the functional profiles of CMV-specific T cells after HSCT, with significant increases in the frequencies of paucifunctional T cells, defined by the number of functions elicited in response to CMV-derived antigen encounter (Figure 4B bottom panels). Taken together, these data show that CMV-specific T cells exhibit a more restricted functional profile as they acquire a more differentiated phenotype after HSCT.
Discussion
In this study, we show that the reconstitution of CMV-specific T-cell immunity in HSCT recipients is, in large part, determined by the specificity and phenotype of the corresponding immune responses in the donor. Importantly, factors that could introduce bias, such as CMV serostatus, graft T-cell dose, preparative regimen, and GVHD prophylaxis, were similar in all 18 donor-recipient pairs studied herein; this enabled us to dissect the impact of immunologic factors in the transfer of CMV-specific T cells to the recipient. In our cohort, the transfer of CMV-specific T cells occurred only with the initial stem cell graft in 8 patients who did not receive a DLI; furthermore, the infusion of donor lymphocytes was not necessarily associated with an increase in CMV-specific T cells at the time of analysis after HSCT. A decrease in the CD4+ T-cell frequency was observed in 3 of 4 recipients who received a DLI; similar decreases were observed for the CMV-specific CD8+ T-cell response in 2 of 2 recipients after DLI. Rather, the successful transfer of T-cell immunity to CMV was determined primarily by the phenotypic characteristics of the CMV-specific T-cell populations resident in the donor. Thus, less differentiated CMV-specific T-cell populations were more likely to persist in the recipient after HSCT; furthermore, although our cohort was small, the presence of a higher frequency of less differentiated CMV-specific T cells in the donor was associated with a decreased incidence of CMV reactivations in the recipient after HSCT. These findings suggest that the persistence of CMV-specific T cells after HSCT-mediated transfer is important for the prevention of CMV reactivation. Of note, the persistence of tumor-specific central memory T cells has been shown to be a key determinant of effective antitumor immunity in murine adoptive transfer experiments39,40 ; more recently, in a nonhuman primate model, it has been shown that adoptively transferred CMV-specific T-cell clones derived from the central memory pool persisted effectively in recipient macaques compared with T-cell clones of the same specificity derived from the effector memory pool.41 Thus, our data suggest that the differentiation status of CMV-specific T cells in the donor determines the proliferative capacity of these cells and their fate after HSCT.
After HSCT, almost all responding T cells exhibited a more differentiated phenotype associated with a restricted functional profile skewed toward the production of proinflammatory cytokines; this is consistent with the recruitment of effector T cells after HSCT, when CMV reactivations were frequent.42,43 Because all patients were receiving low-dose CsA at the time of immunologic analysis after HSCT, it is possible that the immunomodulatory effects of this agent affected our observations at this time.44-47 However, in our cohort, low-dose CsA did not appear to exert adverse effects on either the frequency of the total CMV-specific T-cell response or the secretion of effector cytokines after HSCT because these parameters increased in almost all donor-recipient pairs. Thus, the observed effects on phenotype and function most likely reflect the robust expansion of CMV-specific T cells in immunocompromised CMV-seropositive recipients.
In conclusion, our data show that the fate of CMV-specific T cells that are adoptively transferred during HSCT is determined by the characteristics of the response in the donor. Specific T-cell responses that were less differentiated in the donor tended to persist in the corresponding recipient; conversely, those T-cell responses that were terminally differentiated in the donor either decreased in frequency or were not identified in the recipient. Furthermore, the frequency of CD8+ CMV-specific CD27+ memory T-cell responses in the donor appeared to confer protection from CMV reactivations in the recipient. Additional studies focused on this simple prognosticator of clinical outcome in larger patient cohorts are planned to confirm these findings and determine the extent to which one can predict the likelihood of CMV-specific T-cell persistence and efficacy in the recipient after HSCT from a phenotypic analysis of the corresponding responses in the donor. Several potential ramifications emerge from this work. For example, protocols that investigate the immunization of donors to transfer protective immunity during HSCT might well benefit from vaccination strategies that favor the generation of less differentiated antigen-specific T cells to enhance the establishment of effective immunologic memory in the recipient. Furthermore, clinical adoptive transfer protocols have placed great emphasis on the frequency of IFN-γ–producing T cells for the purposes of immune monitoring; the data presented herein show that this might not be a good predictor of immunologic outcome after HSCT. Thus, overall, our results provide important insights into the factors that influence the successful adoptive transfer of antigen-specific T-cell immunity during HSCT and inform the development of strategies to improve the treatment of tumors and infections in this setting.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, National Heart, Lung and Blood Institute and National Institute of Allergy and Infectious Diseases. D.A.P. is a Medical Research Council (United Kingdom) Senior Clinical Fellow.
National Institutes of Health
Authorship
Contribution: P.S. provided primary conception, execution, and data analysis and drafted the manuscript; J.J.M. and J.M.B. contributed to primary conception, interim discussions, and manuscript preparation; B.J.H. and N.F.H. were involved in execution and interim discussions; P.K.C. and M.R. provided expertise in flow cytometry and panel development and supplied in-house conjugated antibodies; L.J.P. provided the CMV ORF peptides and participated in study design and manuscript preparation; and D.A.P., A.J.B., and D.C.D. were involved in primary conception, data interpretation, interim discussions, and manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daniel C. Douek, Vaccine Research Center, National Institutes of Health, 40 Convent Dr, Bethesda, MD 20892; e-mail: ddouek@mail.nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal