

Abstract
Previously published guidelines for the diagnosis and management of primary immune thrombocytopenia (ITP) require updating largely due to the introduction of new classes of therapeutic agents, and a greater understanding of the disease pathophysiology. However, treatment-related decisions still remain principally dependent on clinical expertise or patient preference rather than high-quality clinical trial evidence. This consensus document aims to report on new data and provide consensus-based recommendations relating to diagnosis and treatment of ITP in adults, in children, and during pregnancy. The inclusion of summary tables within this document, supported by information tables in the online appendices, is intended to aid in clinical decision making.
Introduction
Primary immune thrombocytopenia (ITP) is an acquired immune-mediated disorder characterized by isolated thrombocytopenia, defined as a peripheral blood platelet count less than 100 × 109/L, and the absence of any obvious initiating and/or underlying cause of the thrombocytopenia.1 Until recently, the abbreviation ITP stood for idiopathic thrombocytopenic purpura, but current awareness relating to the immune-mediated nature of the disease, and the absence or minimal signs of bleeding in a large proportion of cases have led to a revision of the terminology.1 Concepts surrounding the mechanisms of thrombocytopenia in ITP have shifted from the traditional view of increased platelet destruction mediated by autoantibodies to more complex mechanisms in which both impaired platelet production and T cell–mediated effects play a role.2-6 Recent epidemiologic data suggest that the incidence in adults is approximately equal for the sexes except in the mid-adult years (30-60 years), when the disease is more prevalent in women.7,8 ITP is classified by duration into newly diagnosed, persistent (3-12 months' duration) and chronic (≥ 12 months' duration).1 Whereas ITP in adults typically has an insidious onset with no preceding viral or other illness and it normally follows a chronic course,9 ITP in children is usually short-lived with at least two-thirds recovering spontaneously within 6 months.10 Signs and symptoms vary widely. Many patients have either no symptoms or minimal bruising, whereas others experience serious bleeding, which may include gastrointestinal hemorrhage (GI), extensive skin and mucosal hemorrhage, or intracranial hemorrhage (ICH). The severity of thrombocytopenia correlates to some extent but not completely with the bleeding risk.7,11 Additional factors (eg, age, lifestyle factors, uremia) affect the risk and should be evaluated before the appropriate management is determined.
The investigation and management of ITP patients vary widely. The purpose of this consensus document is to comment on new data and provide recommendations relating to diagnosis and treatment. Final judgment regarding care of individual patients should, however, lie with the responsible health care professional and be based on careful investigation of individual circumstances.
Methods
Composition of the panel.
The panel included 22 members with recognized clinical and research expertise in ITP representing North America (United States, 7; Canada, 1), Europe (France, 1; Italy, 2; Spain, 1; Switzerland, 1; United Kingdom, 8), and Australia (1).
Assessment of the literature.
Articles were identified by a computer-assisted search of the literature published in English using the National Library of Medicine PubMed database. The search criteria were: ‘immune thrombocytopenic purpura’, ‘idiopathic thrombocytopenic purpura’, ‘ITP’, and ‘autoimmune thrombocytopenic purpura’. A subsequent search was performed using the corresponding MedLine MeSH terms and cross-referenced with the original search to consolidate the primary results. Abstracts from the European Haematology Association (EHA), American Society of Hematology (ASH), and International Society on Thrombosis and Haemostasis (ISTH) annual meetings from 2003 to 2007 were also reviewed for relevance. Papers were graded using a specific scoring system (supplemental Document 1). Levels of evidence were reviewed by the lead writing committee at 2 face-to-face meetings throughout the manuscript preparation, and all authors were given the chance to dispute the levels assigned at each review stage. Randomized controlled trials (RCTs) were graded as providing the highest level of evidence, with case studies and expert opinion the lowest. Grades of recommendation were based on the supporting evidence levels.
The relevant data are presented in the text with additional supporting tables and appendices available on the Blood website (see the Supplemental Materials link at the top of the online article).
Diagnostic approach in patients with suspected ITP
Diagnostic tools for adults and children with suspected ITP were grouped into 3 sections of recommendation (supplemental Document 8, Recommendation Box 1; Table 1). A presumptive diagnosis of ITP is made when the history, physical examination, complete blood count, and examination of the peripheral blood smear do not suggest other etiologies for the thrombocytopenia. There is no “gold standard” test that can reliably establish the diagnosis. Response to ITP-specific therapy, for example, intravenous immunoglobulin (IVIg) and intravenous anti-D, is supportive of the diagnosis, but a response does not exclude secondary ITP.
Recommendations for the diagnosis of ITP in children and adults
| Basic evaluation | Tests of potential utility in the management of an ITP patient | Tests of unproven or uncertain benefit |
|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
| ||
| ||
| ||
| ||
|
| Basic evaluation | Tests of potential utility in the management of an ITP patient | Tests of unproven or uncertain benefit |
|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
| ||
| ||
| ||
| ||
|
Refer also to supplemental Document 2.
Rh indicates rhesus; H pylori, Helicobacter pylori; HIV, human immunodeficiency virus; HCV, hepatitis C virus; PCR, polymerase chain reaction; CMV, cytomegalovirus; TPO, thrombopoietin; and PaIgG, platelet-associated immunoglobulin G.
Quantitative immunoglobulin level measurement should be considered in children with ITP and is recommended in those children with persistent or chronic ITP as part of the reassessment evaluation.
Recommended by the majority of the panel for adult patients regardless of geographic locale.
Patient history
Thrombocytopenia can be caused by myriad conditions including systemic disease, infection, drugs, and primary hematologic disorders (Table 2). In approximately 60% of pediatric cases, there is a history of a previous infection.12 An increased risk of ITP is also associated with measles-mumps-rubella vaccination.13 Bleeding after previous surgery, dentistry, and trauma should be considered when estimating the possible duration of chronic thrombocytopenia or an alternative bleeding disorder. If a diagnosis of ITP is established, contraindications to or cautions about corticosteroid therapy should be noted. Inherited thrombocytopenia should be considered in patients with long-standing thrombocytopenia unaffected by treatment and in those with a family history of thrombocytopenia or bleeding disorders.
Frequent examples of differential diagnosis of ITP and potential alternative causes of thrombocytopenia identified by patient history
|
|
Refer also to supplemental Document 2.
The possibility of abuse must be considered by emergency department staff when dealing with a young child who presents with bruising and purpura for the first time (evidence level IV). Children with infections such as meningococcal sepsis usually have other systemic features that help rapidly differentiate such conditions from ITP.
Physical examination
Physical examination should be normal aside from bleeding manifestations. Mild splenomegaly may be found in younger patients, but moderate or massive splenomegaly suggests an alternative cause. Constitutional symptoms, such as fever or weight loss, hepatomegaly, or lymphadenopathy might indicate underlying disorder such as HIV, systemic lupus erythematosus (SLE), or a lymphoproliferative disease.
Peripheral blood count
ITP is characterized by isolated thrombocytopenia with an otherwise normal complete blood count. Anemia from blood loss may be present, but it should be proportional to the amount, and the duration, of bleeding and may result in iron deficiency (evidence level IV). If anemia is found, the reticulocyte count may help define whether it the result of poor production or increased destruction of red blood cells (RBCs).
Evaluation of peripheral blood smear
Evaluation of the peripheral blood smear by a qualified hematologist or pathologist is paramount to the diagnosis of ITP. This may demonstrate abnormalities that are not consistent with ITP, such as schistocytes in patients with thrombotic thrombocytopenic purpura–hemolytic uremic syndrome, or leukocyte inclusion bodies in MYH9-related disease. Excessive numbers of giant or small platelets may indicate an inherited thrombocytopenia (supplemental Document 9). Pseudo-thrombocytopenia due to ethylenediaminetetra acetic acid (EDTA)–dependent platelet agglutination should also be excluded14 (evidence level III).
Bone marrow examination
Bone marrow examination may be informative in patients older than 60 years of age, in those with systemic symptoms or abnormal signs, or in some cases in which splenectomy is considered.15-18 Both a bone marrow aspirate and a biopsy should be performed. In addition to the morphologic assessment, flow cytometry and cytogenetic testing should be considered (evidence level IIb-IV). Flow cytometry may be particularly helpful in identifying patients with ITP secondary to chronic lymphocytic leukemia (CLL).19
Helicobacter pylori testing
The detection of H pylori infection, preferably with the urea breath test or the stool antigen test, should be considered in the work-up of adults with typical ITP where it may have clinical impact20 (evidence level IIa). Serologic detection may be used but is less sensitive and less specific than the other tests; furthermore, the test may produce false positive results after IVIg therapy. Except in high-prevalence areas, the literature does not support routine testing in children with ITP.
HIV and HCV testing
The thrombocytopenia associated with HIV and hepatitis C virus (HCV) infections may be clinically indistinguishable from primary ITP and can occur several years before patients develop other symptoms.21 Routine serologic evaluation for HIV and/or HCV infection in adult patients with suspected ITP, regardless of local background prevalence and personal risk factors documented in the patient history, is recommended. Control of these infections may result in complete hematologic remission (evidence level IIa).21
Quantitative immunoglobulin level testing
Baseline immunoglobulin (Ig) levels (IgG, IgA, and IgM) should be measured in adults (evidence level IV). They should also be considered at baseline in children with ITP, and measured in those children with persistent or chronic ITP as part of a reassessment evaluation. Low levels may reveal conditions such as common variable immunodeficiency (CVID) or selective IgA deficiency. Treatment of ITP with immunosuppressive agents is therefore relatively contraindicated in CVID. Although Ig levels should ideally be tested prior to use of IVIg, it will often be necessary to treat the patient before the results are known (evidence level IV).
Direct antiglobulin test
A positive direct antiglobulin test (DAT) was found in 22% of 205 patients (19 children, 186 adults) with ITP,22 but its clinical significance is unknown. A DAT is generally appropriate if anemia associated with a high reticulocyte count is found and if treatment with anti-D immunoglobulin is being considered.
Blood group Rh(D) typing
This is important if anti-D immunoglobulin is being considered.
Tests of potential utility
Antiplatelet antibody assays: glycoprotein-specific antibody testing.
Antiphospholipid antibodies.
Antiphospholipid antibodies (APLA), including anticardiolipin antibodies and lupus anticoagulant, can be found in approximately 40% of otherwise typical adult patients with ITP.25 The presence of APLA does not appear to affect the response to ITP treatment. Routine testing is not recommended in the absence of symptoms of antiphospholipid syndrome.
Antinuclear antibodies.
A positive antinuclear antibody (ANA) test may be a predictor of chronicity in childhood ITP26 (evidence level IIb).
Antithyroid antibody and thyroid function testing.
Eight percent to 14% of ITP patients followed longitudinally developed clinical hyperthyroidism.27 Others developed antibodies to thyroglobulin and may eventually develop hyper- or hypothyroidism. Mild thrombocytopenia has been reported in patients with hyperthyroidism (reduced platelet survival) and hypothyroidism (possible decreased platelet production), which often resolve with restoration of the euthyroid state. It may also be useful to measure antibodies to thyroglobulin and thyroid-stimulating hormone (TSH) to identify patients at risk for clinical thyroid disease.
Testing for other acute and persistent infections.
Acute viral infections and some vaccinations (with live attenuated virus) have been associated with thrombocytopenia, which is usually transient. Some chronic infections, for example, parvovirus and cytomegalovirus (CMV), can also produce thrombocytopenia.
Diagnostic tests of unproven or uncertain benefit
Several other tests (Table 1) currently have no proven role in the differential diagnosis of ITP from other thrombocytopenias and do not guide patient management.
Management of adult ITP
Although RCT data are now available for some new ITP treatments (eg, romiplostim, eltrombopag), only a limited number of RCTs have been conducted in adults using traditional therapies and even fewer for other agents. Whereas this document gives a widespread approach to treatment (Table 3), a general rule is that treatment should always be tailored to the individual patient. All treatment options are listed alphabetically so as to show no preference for a particular therapy. Response rate criteria vary between studies, making direct comparisons of response rates given for individual treatment options difficult.
Therapies for the treatment of ITP
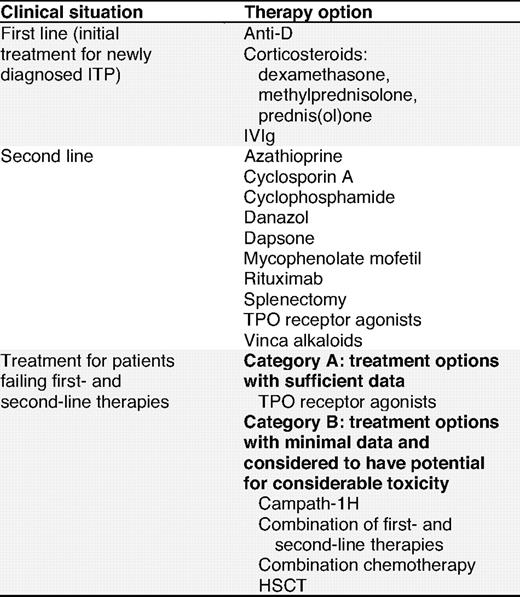
Treatment options for ITP are listed in alphabetical order and thus do not imply a preferred treatment option.
HSCT indicates hematopoietic stem cell transplantation; TPO, thrombopoietin; and IVIg, intravenous immunoglobulin.
Due to the costs of modern drug development, newer therapies may be expensive, which could potentially limit their availability and use in some countries. Financial resources, either of the individual patient or of the publicly funded health care system, may also strongly impact the choice of treatment. However, this higher cost needs to be offset by the fact that these new agents are not immunosuppressive, have undergone rigorous randomized controlled clinical studies, and appear to have high efficacy.
Who should be treated?
Relevant factors that contribute to management decisions include the extent of bleeding, comorbidities predisposing to bleeding, complications of specific therapies, activity and lifestyle, tolerance of side effects, potential interventions that may cause bleeding, accessibility of care, patient expectations, patient worry or anxiety about disease burden, and patient need for non-ITP medications that may create a bleeding risk.
Although hemorrhagic death is a major concern, analysis of data from 17 adult case studies estimated the rate of fatal hemorrhage to be 0.0162 to 0.0389 cases per adult patient-year at risk.28 Patients older than 60 years and those with previous hemorrhage have a higher bleeding risk.29 Bleeding and infection contribute equally to mortality.30
Treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of the following: bleeding due to platelet dysfunction or another hemostatic defect, trauma, surgery,31 clearly identified comorbidities for bleeding, mandated anticoagulation therapy, or in persons whose profession or lifestyle predisposes them to trauma. Patient preference must also be considered when discussing treatment options. Detailed consensus-based recommendations regarding target platelet counts during surgery in adults were provided previously16 and have been further modified (supplemental Document 8, Recommendation Box 2).
First-line treatment: initial treatment for newly diagnosed patients
Response rate criteria vary between studies (supplemental Document 8, Recommendation Box 3) and it is not possible to compare response rates for individual treatment options.
Corticosteroid therapy.
Corticosteroids are the standard initial treatment. Additionally, they may also reduce bleeding, independent of the platelet count rise by means of a direct effect on blood vessels.32,33 Unfortunately their adverse effects rapidly become apparent and create significant complications. With time, the detrimental effects of corticosteroids often outweigh their benefits. Prednisone is the standard initial first-line therapy for ITP patients.22,34,35 Prednisone is usually given at 0.5 to 2 mg/kg/d until the platelet count increases (≥ 30-50 × 109/L), which may require several days to several weeks.9,36,37 Although the treatment is effective, patients are at risk of developing corticosteroid-related complications that vary with the dose and duration. To avoid corticosteroid-related complications, prednisone should be rapidly tapered and usually stopped in responders, and especially in non-responders after 4 weeks.36,38
Dexamethasone.
Although it has been abandoned in the treatment of chronic refractory ITP patients,39,40 recent results from 2 large first-line studies with dexamethasone suggest both a high initial response rate and a substantial sustained response rate (Table 4). Administration of dexamethasone 40 mg/day for 4 days (equivalent to ∼ 400 mg of prednisone per day) produced sustained response in 50% of newly diagnosed adults with ITP. In another study, 4 cycles of dexamethasone (40 mg/day for 4 days) given every 14 days produced an 86% response rate with 74% having responses lasting a median time of 8 months.41 RCTs are needed to definitively assess these encouraging results and to distinguish whether pulse dexamethasone is the preferred corticosteroid approach with regard to response, duration of response, and toxicity.
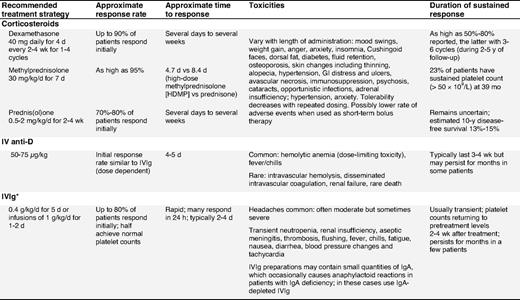
Methylprednisolone.
Parenteral administration of high-dose methylprednisolone has been used in various regimens to treat patients failing first-line therapies,42,43 with 80% response rates. Due to the short-term responses to methylprednisolone, maintenance therapy with oral corticosteroids may be required (evidence level IV).
Intravenous anti-D (IV anti-D).
IV anti-D is appropriate for Rh(D) positive, non-splenectomized ITP patients. It should be avoided in those with autoimmune hemolytic anemia, to avoid exacerbation of hemolysis. Blood group, DAT, and reticulocyte count are required before treating with IV anti-D.44,45
IV anti-D may be an effective alternative to IVIg, as it can be infused in a shorter time, is produced from a smaller donor pool, has a potentially longer response,46,47 and may reduce the need for splenectomy.48-50 Two studies have demonstrated that IV anti-D administered at 75 μg/kg, instead of the licensed 50 μg/kg dose, increases the overall platelet count comparable to that of IVIg.51,52 Premedication with paracetamol/acetaminophen, or corticosteroids (eg, 20 mg of prednisone), is recommended to reduce the risk of fever/chill reactions especially with the higher dose.51 Mild anemia is expected and may be dose-limiting.48 Rare, but very serious, even fatal, cases of intravascular hemolysis, disseminated intravascular coagulation, and renal failure have been reported44,45,53 (evidence level Ib-III). IV anti-D is a pooled, biological blood product, the risks of which must be explained to patients. Recent safety concerns have been raised regarding the product WinRho SDF that has prompted its removal from the European market. Until the full nature of these adverse effects is assessed, this agent should be used with caution. A more convenient54-57 and perhaps more tolerable56,57 delivery of anti-D immune globulin by the intramuscular54,55 or subcutaneous56,57 route has been reported in limited, open-label trials of adults54 and children55-57 with chronic ITP. In all, the majority of patients exhibited a platelet response within 1 week of treatment, with no reports of severe adverse events. Controlled prospective trials are needed to place subcutaneous or intramuscular anti-D among recommended therapies for ITP.
IVIg.
Numerous controlled trials have been performed with high-dose IVIg since its initial use approximately 20 years ago58 and have shown initial response rates comparable to those of corticosteroids but with a shorter time to response.16 ITP patients with CVID may be treated with high-dose IVIg followed by maintenance dosing of 0.3 to 0.4 g/kg every 3 to 4 weeks.59
Although IVIg is associated with higher toxicity, especially headaches, and the need for a prolonged infusion (over at least several hours), IVIg recipients are more likely to attain a platelet increase within 24 hours at a dose of 1 g/kg (1-2 infusions over 2 days) compared with the historical treatment regimen (0.4 g/kg/d over 5 days).60 Rare but serious toxicities include renal failure and thrombosis.61,62 The fear of transmission of infectious disease persists, but there is no recent evidence for transmission of HIV, HCV, and HBV, and human T-cell lymphotrophic virus type 1 (HTLV-1). It appears that in some patients corticosteroids may enhance the IVIg response. In addition to this, the concomitant use of corticosteroids may reduce infusion reactions and prevent aseptic meningitis.
Emergency treatments
An urgent increase in platelet count may be required for some thrombocytopenic patients needing surgical procedures, at high risk of bleeding, or with active central nervous system (CNS), GI, or genitourinary bleeding (supplemental Document 8, Recommendation Box 4).
Although changing from corticosteroids to IVIg or anti-D may be effective in emergency settings, combining first-line therapies is appropriate: prednisone and IVIg are recommended for the emergency treatment of patients with uncontrolled bleeding. High-dose methylprednisolone (HDMP) may also be useful in this setting. Other therapies that work rapidly include platelet transfusion, possibly in combination with IVIg, and emergency splenectomy.63 There is also some evidence of rapid response to vinca alkaloids.
General measures.
These include cessation of drugs reducing platelet function, control of blood pressure, inhibition of menses, and efforts to minimize trauma (evidence level IV). However, there may be instances in which oral anti-coagulation or antiplatelet medication is necessary (eg, in patients with cardiac stents requiring aspirin and/or clopidogrel) and necessitates raising the threshold platelet count for treatment. In patients with reduced renal function, hemostasis may be improved with desmopressin and by maintaining hemoglobin at a minimum of 10 g/dL.
Platelet transfusions with or without IVIg.
Platelet transfusion increases the posttransfusion platelet count by more than 20 × 109/L in 42% of bleeding ITP patients and may reduce bleeding.64 In a retrospective study of 40 patients63 (evidence level IIb), concurrent administration of platelet transfusions and IVIg was associated with resolution of bleeding, rapid restoration of adequate platelet counts, and minimal side effects (evidence level III/IV).
Vinca alkaloids.
As a single agent, vincristine induces a platelet count increase in a small fraction of chronic ITP patients (evidence level IV). However, when combined with other agents it may be a useful approach in patients requiring emergency treatment65 (evidence level IIb).
Emergency splenectomy.
See “Splenectomy” in “Second-line therapy: surgical.”
Antifibrinolytics.
Antifibrinolytic agents, such as oral or IV tranexamic acid and episilon-aminocaproic acid may be useful in preventing recurrent bleeding in patients with severe thrombocytopenia; however, the efficacy has not been evaluated by randomized trial in ITP patients. Tranexamic acid (1 g, 3 times daily orally) and episilon-aminocaproic acid (1-4 g every 4-6 hours [maximum dose, 24 g/d]) may be of special value in certain dental or surgical procedures.
Emergency treatments that are not indicated
Plasmapheresis.
Plasmapheresis has been studied in small cohorts of ITP patients, some of whom had acute ITP. No patients with chronic ITP showed a response66 (evidence level III).
Second-line treatment options for adult ITP patients
Splenectomy and a large number of drugs have been used as second-line therapy with variable success. Physicians are required to make individual judgments about the nature of second-line treatment based on bleeding history, comorbidities, patient expectations, and compliance.
The main goal of second-line therapy is to attain a sustained increase of the platelet count that is considered hemostatic for the individual patient. Available treatment modalities have quite different mechanisms of action and can be broadly categorized into those that are given only once (or for only one course) and are intended to induce long-term remission (splenectomy, rituximab), and those that need continued or chronic administration (corticosteroids, immunosuppressive agents, thrombopoietin receptor agonists).
Depending on the clinical setting, splenectomy is deferred in most patients for at least 6 months. This may be due to patient preference or other active comorbidities and to the understanding that spontaneous improvement or late remission may occur 6 to 12 months after diagnosis; indeed, some patients may spontaneously remit even years after diagnosis.
Second-line therapy: medical.
Treatment options are listed alphabetically so as to show no preference for a particular therapy (supplemental Document 8, Recommendation Box 5; Table 5).
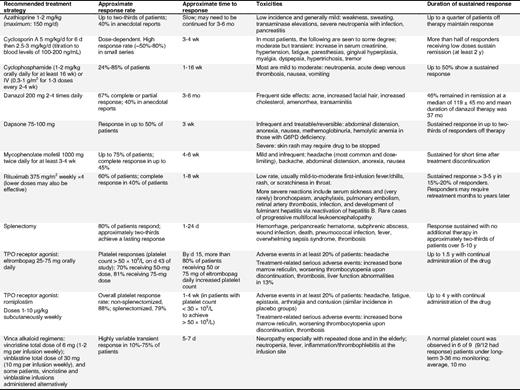
Azathioprine.
Despite few new data, consensus was that this agent is still useful. Investigators have reported complete responses in 45% of 53 patients (40 splenectomized) treated with azathioprine (150 mg/day) for a median of 18 months.67 Although continued therapy is generally required, often a reduced dose suffices. Leukemia has only rarely been associated with azathioprine in other disorders but has not been described in ITP patients68 (evidence level III).
Cyclosporin A.
Cyclosporin A (2.5-3 mg/kg/d) is effective as a single agent in ITP patients or when given with prednisone, but its side effects may make it unsuitable for some patients (eg, older patients and those with renal insufficiency). Clinical improvement was observed in more than 80% of patients resistant to first-line therapy, with 42% achieving a complete response69 (evidence level IIa). Remissions may be durable (mean, 29 months) following discontinuation of treatment70 (evidence level IIb). Side effects are usually moderate but transient and include fatigue, renal insufficiency, hypertension, and neuropathy.69
Cyclophosphamide.
Immunosuppression with cyclophosphamide, either orally (1-2 mg/kg daily for at least 16 weeks) or intravenously (0.3-1 g/m2 for 1-3 doses every 2-4 weeks), has been used for patients refractory to corticosteroids and/or splenectomy. Response rates varied from 24% to 85%43,71,72 and toxicity was mild to moderate.72 However, there are reports of ITP and SLE patients developing acute myeloid leukemia (AML) after cyclophosphamide therapy.73 The complication of sterility after treatment for ITP has not been adequately addressed.
Danazol.
Danazol is an attenuated androgen administered orally at a dose of 200 mg, 2 to 4 times daily (10-15 mg/kg/d). Response rates of 60% to 67% have been recorded (> 50 × 109/L for ≥ 2 months in 57 ITP patients after splenectomy).74 Older females and splenectomized patients may have the highest rate of response.74
Dapsone.
Dapsone is a moderate corticosteroid-sparing agent that is usually administered orally at a dose of 75 to 100 mg/d.75 Dapsone may delay splenectomy for up to 32 months in patients who have not responded to first-line corticosteroid therapy (evidence level IIb). However, splenectomized patients have a low response rate.76
Male patients at risk for glucose-6-phosphate dehydrogenase (G6PD) deficiency should be screened before starting treatment and monitored for hemolysis and methemoglobinemia77 (evidence level III).
Mycophenolate mofetil.
Mycophenolate mofetil (MMF), an antiproliferative immunosuppressant, has been shown to be useful in some ITP patients. Administration of MMF at progressively increasing doses (250 mg up to optimally 1000 mg/day twice a week over 3 weeks) produced platelet increase in 39% of patients with refractory ITP, but was not sustained78 (evidence level IIb). In a retrospective study, the overall response rate was 78% (major response, > 80 × 109/L; and moderate response, 30-80 × 109/L at 3 months).79
Rituximab.
Several publications have reported the use of rituximab in ITP patients since previous consensus documents were issued15,16 and suggest that about 60% of patients respond, with approximately 40% achieving complete response.80 Responses generally occur after 1 to 2 weeks to 6 to 8 weeks81,82 and last from 2 months in partial responders to 5 years or longer in 15% to 20% of initially treated patients.80 Most patients with a durable (> 1 year) complete response will respond to repeat treatment if they relapse (evidence level IIa-III).
After 2 years of observation in a prospective, open-label, single-arm phase 2 trial, 33% of patients had a platelet count of 50 × 109/L or higher and 40% had a platelet count of 30 × 109/L or higher without any additional treatment.83 Although the studies presented used rituximab doses of 375 mg/m2, lower doses (100 mg IV weekly for 4 weeks) may also be effective, although associated with a longer time to response.84 At the current time the standard dose of rituximab for ITP patients is unknown, and, due to the potential toxicity and expense of the agent, future studies are required to identify the optimal dose. High response rates have recently been reported for a combination of rituximab with high-dose dexamethasone as initial therapy.85
Rituximab is contraindicated in patients with evidence of active hepatitis B infection (eg, positive hepatitis B/C core antibody). Adverse events associated with rituximab are usually mild or moderate, with a low incidence of infections.82,86 There are also reports of more than 50 cases of progressive multifocal leukoencephalopathy associated with rituximab treatment in patients with lymphoma and more recently a limited number of patients with SLE and ITP.87,88 Hence, additional long-term safety data are required. These cases tend to occur in patients who are heavily immunosuppressed and on combination treatments.
Thrombopoietin-receptor agonists: romiplostim and eltrombopag.
Rather than modulating the immune system, another therapeutic approach is to stimulate platelet production. Thrombopoietin (TPO) is the primary factor regulating platelet production,89 and several TPO-receptor agonists have been developed that activate the TPO receptor and increase platelet production.90-93 Two agents, romiplostim and eltrombopag, are FDA-approved for the treatment of ITP. Romiplostim is administered as a 1 to 10 μg/kg subcutaneous weekly injection.93,94 Eltrombopag is an oral non-peptide TPO-receptor agonist administered as a 25, 50, or 75 mg daily dose92,94,95 (evidence level Ib/IIa).
Data from phase 1-3 trials have demonstrated that both drugs are highly effective in increasing the platelet count in both healthy volunteers and ITP patients.90,92-98 In 2 parallel, placebo-controlled, double-blind randomized phase 3 trials, romiplostim was given to 63 splenectomized and 62 non-splenectomized patients for 6 months.93 An overall platelet response rate (≥ 4 out of 24 study weeks > 50 × 109/L) was observed in 79% and 88% of romiplostim-treated patients, compared with 0% and 14% in the respective placebo arms.93 Similar results have been achieved with eltrombopag in chronic relapsed or refractory ITP patients (n = 114); 59% of eltrombopag-treated patients compared with 16% of placebo-treated patients achieved a platelet count of 50 × 109/L or higher on day 43 of the study.94
Across the 2 romiplostim studies, 87% of romiplostim-treated patients reduced or discontinued concurrent ITP therapy, including corticosteroids and IVIg.93 Long-term data with romiplostim showed that responses were sustained for up to 4 years on continuous therapy, with most patients able to decrease or discontinue concurrent corticosteroid therapy.98 This is an important finding, especially as it affects patients who may have been on immunosuppressive treatment for a long period of time. TPO-receptor agonists have the potential to minimize morbidity and mortality in these patients.
Although most adverse effects were mild, concerns have been raised over the increased bone reticulin found in 10 of more than 271 patients included in the romiplostim trials and in 7 of 117 patients in the eltrombopag trials. Long-term studies will address the importance of this finding and determine whether routine monitoring is required.93,98 Although reported in rodent studies with eltrombopag, there was no increase in cataracts observed in the ITP studies.92,94,99 Liver function test abnormalities were seen in 13% of eltrombopag-treated patients.100
Due to their mechanism of action, TPO-receptor agonists are considered a maintenance therapy. Upon cessation of treatment, most patients return to lower platelet counts (∼ 10% transiently falling below baseline platelet counts); however, a few patients are able to discontinue treatment successfully.93
Vinca alkaloids.
Second-line therapy: surgical.
Splenectomy.
Eighty percent of patients respond to splenectomy, and response is sustained in 66% with no additional therapy for at least 5 years103-105 (supplemental Document 8, Recommendation Box 6). Many patients without a complete response can still expect a partial or transient response.15,106 Approximately 14% of patients do not respond and approximately 20% of responders relapse weeks, months, or years later103 (evidence level IIb).
Complications of splenectomy.
Complications of splenectomy include bleeding, infection, thrombosis, prolonged hospitalization, readmission to the hospital, and requirement for additional intervention.104 Reported complication rates vary considerably30,103,104,107,108 and may be greater in patients aged 65 years or older.29 In a recent systematic analysis, splenectomy complication rates were 12.9% with laparotomy and 9.6% with laparoscopy; mortality was 1.0% with laparotomy and 0.2% with laparoscopy.104
Predicting response to splenectomy.
There is no widely accepted test predicting response to splenectomy. Response to oral corticosteroids or high-dose IVIg has a low predictive value110,111 (evidence level IIb). Indium-labeled autologous platelet scanning may be the most sensitive predictor of response to splenectomy, but here too studies vary.112,113 When scanning reveals splenic platelet destruction, approximately 90% of patients respond to splenectomy.112 This test is currently limited to several research centers, but if available may be useful before splenectomy (evidence level III, grade B recommendation).
Accessory splenic tissue (evidence level III/IV).
Imaging techniques show accessory splenic tissue in up to 12% of splenectomized patients and almost all is removed during surgery.114 In patients who relapse following an initial response to splenectomy, assessment for accessory spleen should be considered. However, in patients who never responded to initial splenectomy, response is extremely rare.42,115
Prevention of infection after splenectomy.
Splenectomized patients are at lifelong risk for uncontrolled infection with a poor outcome from Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae.116,117
Patients are usually given prophylactic polyvalent pneumococcal, meningococcal C conjugate, and H influenzae b (Hib) vaccines at least 4 weeks before (preferably) or 2 weeks after splenectomy and revaccinated according to the country-specific recommendations (evidence level IV).118,119 In patients who have received rituximab in the previous 6 months, vaccinations may not be effective. Vaccination for these patients should be readdressed once B-cell recovery has occurred.
In some studies, asplenic patients were given long-term antibiotic prophylaxis (phenoxymethylpenicillin 250-500 mg twice a day or equivalent, or erythromycin 500 mg twice a day).120 However, the benefit of lifelong antibiotic prophylaxis is unproven121,122 and the risk of late infection is quite low, and therefore no consensus has been reached.123
A practical policy is for splenectomized patients to have a home supply of antibiotics (eg, penicillin VK, erythromycin, or levofloxacillin) for use in case of a febrile illness. Patients should be educated about the risk of postsplenectomy infection, including the need to go to the emergency department if fever higher than 101°F (38°C) occurs. In addition, cards should be carried to alert physicians that the patient is asplenic; some patients may wish to wear alert bracelets or pendants (grade C recommendation, evidence level IV).
Treatment options for adult patients failing first- and second-line therapies
Patients failing first- and second-line therapies.
Approximately 20% of patients do not attain a hemostatic platelet count after splenectomy or after first- and second-line medical approaches (supplemental Document 8, Recommendation Box 7); an additional 10% to 20% of splenectomy responders eventually relapse (evidence level IV). These patients may be able to tolerate severe thrombocytopenia (ie, platelet counts as low as 10 × 109/L) relatively well with near-normal quality of life (QoL). However, some have consistent and statistically significant deficits on QoL measures, bleeding, and increased risk of death.28,124-126 For those who fail standard therapies and still require treatment, options are limited. In this situation, the risk of further therapy must be discussed with the patient and compared with the benefit of that therapy. In addition, other potential etiologies for thrombocytopenia should be exhaustively explored.127 Some patients opt to live with lower platelet counts instead of undergoing toxic treatments (Table 6).
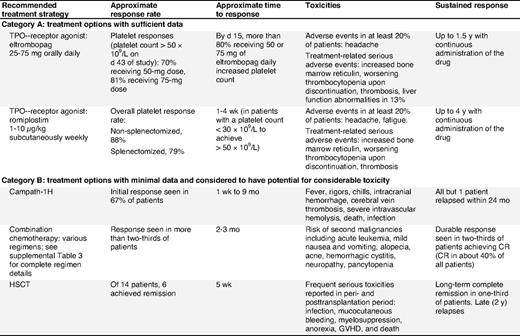
Combination chemotherapy.
Combination chemotherapy may be effective for some chronic refractory ITP patients.65,128,129 A combination of cyclophosphamide (100-200 mg/d IV) on days 1 through 5 or 7 and prednisone (0.5-1.0 mg/kg orally daily) on days 1 through 7, combined with vincristine (1-2 mg IV) on day 1, and one of the following: azathioprine (100 mg orally daily) on days 1 through 5 or 7 or etoposide (50 mg orally daily) on days 1 through 7 has been evaluated. The overall response rate in the 31 patients was 68% including a complete response in 42%; therapy was well tolerated (evidence level IIb). Longer-term follow-up is required to assess durability of remission and the risk of secondary cancers.
Campath-1H.
Campath-1H is an alternative therapeutic option for severe, refractory ITP130 ; however, this drug has the potential to cause severe, possibly life-threatening, immunosuppression and usually requires prolonged antifungal, antibacterial, and antiviral prophylaxis.
Hematopoietic stem cell transplantation.
Remissions have been induced in some patients with chronic refractory ITP using autologous or allogeneic hematopoietic stem cell transplantation (HSCT)131-134 (evidence level IIb/III). However, potentially fatal toxicities such as neutropenic fever, cerebral hemorrhage, and septicemia may occur.133 HSCT is warranted only in patients with severe chronic refractory ITP with bleeding complications unresponsive to other modalities. However, very few long-term responses have been recorded.
TPO-receptor agonists: romiplostim and eltrombopag.
TPO-receptor agonists have been studied in patients after splenectomy and show an approximately 79% overall response rate90,92-94,98 (evidence level Ib). TPO-receptor agonists may be a costly option, and thrombocytopenia will usually return upon cessation of treatment. However, these are the only treatments for refractory ITP that have been shown to be effective in RCTs. In view of the apparent low toxicity and good tolerability of these treatments, many patients would choose to be on them indefinitely. To date, administration of romiplostim has been continued for up to 4 years without loss of benefit or cumulative toxicity.98
Therapies whose use is not justified
The following approaches have been reviewed and are not justified for use in ITP due to evidence affirming lack of efficacy or excessive toxicity: colchicine, interferon α, protein A immunoadsorption column, plasmapheresis as an isolated approach, vitamin C, recombinant factor VIIa.
Supportive care
Antifibrinolytics.
See “Emergency treatments” (evidence level IV).
Inhibition of menstrual bleeding.
Both progesterone-containing intrauterine contraceptive device and oral contraceptives can decrease the frequency and amount of menstrual bleeding135 (evidence level IIb).
Other measures.
Patient groups (such as the ITP Support Association [http://www.itpsupport.org.uk/], the Platelet Disorder Support Association [http://www.pdsa.org/], and the ITP Foundation [http://www.itpfoundation.org]) offer psychological support to patients by providing relevant information about their condition, available treatments, and advice on how to live with ITP.
Thrombocytopenia in pregnancy
Presentation of thrombocytopenia during pregnancy
Platelet counts are usually lower in pregnant than in non-pregnant women.132 This gestational thrombocytopenia is caused by a combination of hemodilution and increased platelet activation and clearance. A decrease of approximately 10% in platelet count is typical toward the end of the third trimester. ITP is estimated to occur in 1 in 1000 to 1 in 10 000 pregnant women.137 Women with previously diagnosed ITP may experience exacerbation or relapse.138,139
A study of 119 pregnancies in 92 women with ITP found that 31% required intervention. Pregnancy is associated with a procoagulant state in preparation for the hemostatic challenge of delivery140 due to increased levels of fibrinogen, factor VIII, and von Willebrand factor; suppressed fibrinolysis; and a reduction in the activity of protein S.140 These changes may result in fewer bleeding symptoms and therefore a greater tolerance to ITP in pregnant compared with non-pregnant women (evidence level IV).
Diagnosis of ITP first presenting in pregnancy
Recommendations for the diagnosis and management of ITP in pregnancy are mainly based on clinical experience and expert consensus, as there are few RCTs.
The diagnosis of ITP involves the exclusion of other causes of thrombocytopenia during pregnancy. As in the non-pregnant setting, diagnostic tests are used to exclude alternative causes of thrombocytopenia (see Tables 1–2 and supplemental Document 8). The work-up of a pregnant ITP patient is essentially the same as that of a non-pregnant patient; taking into account the following additions and exceptions: gestational thrombocytopenia, preeclampsia, HELLP syndrome, DIC, folate deficiency, massive obstetrical hemorrhage, acute fatty liver, antiphospholipid antibody syndrome.
Laboratory investigations for the diagnosis of ITP in pregnancy.
Bone marrow examination is not required to make the diagnosis of ITP in pregnancy (supplemental Document 8, Recommendation Box 8). The measurement of maternal antiplatelet immunoglobulin has no value in the routine diagnosis of ITP in pregnancy.
Management of ITP in pregnancy
Optimum management of ITP in pregnancy requires collaboration among the obstetrician experienced in the management of ITP, the hematologist, the obstetric anesthetist, and the neonatologist. Treatment is largely based on the risk of maternal hemorrhage. Studies have shown that pregnancy in women with ITP can proceed safely with low hemorrhagic risk for both infants and mothers.138,141,142 Because platelet counts may fall in the third trimester, the frequency of platelet count measurement increases to assist in making decisions regarding delivery. The aim of peripartum treatment is to ensure that there is a satisfactory maternal platelet count for delivery.
Target platelet counts for treatment.
Throughout the first 2 trimesters, treatment is initiated (1) when the patient is symptomatic, (2) when platelet counts fall below 20 to 30 × 109/L, or (3) to produce an increase in platelet count to a level considered safe for procedures. Patients with platelet counts at 20 to 30 × 109/L or higher do not routinely require treatment. They should be monitored more closely as delivery approaches.143
The lowest platelet count at which it is safe to administer spinal or epidural anesthesia remains controversial due to the theoretical risk of epidural hematoma formation and neurological damage.144-146 Obstetric anesthetists generally recommend a platelet count of at least 75 × 109/L to allow administration of spinal or epidural anesthesia. Hematologists believe that a platelet count of at least 50 × 109/L is adequate to allow for cesarean section.
Recommended treatment options for the management of ITP in pregnancy
Primary treatment options for maternal ITP are similar to those of other adult ITP patients (supplemental Document 8, Recommendation Box 9). Corticosteroids and IVIg are the first-line treatments for maternal ITP.143 The limited evidence for the use of IV anti-D,147,148 splenectomy, and azathioprine149-151 is presented in the subsections that follow. Vinca alkaloids, rituximab, danazol, TPO-receptor agonists, and most immunosuppressive drugs (other than azathioprine) should not be used during pregnancy because of possible teratogenicity.
First-line treatment: initial treatment for newly diagnosed patients.
Corticosteroids.
Prednisone at a low dose (10-20 mg/d) is given initially and then adjusted to the minimum dose that produces a hemostatically effective platelet count. Worsening of the thrombocytopenia may complicate the last weeks before delivery so tapering should not be pursued aggressively at that time. Short-term, low-dose prednisone is generally considered to be effective and safe for the mother, but corticosteroids can exacerbate hypertension, hyperglycemia, and osteoporosis and may cause excessive weight gain and psychosis. After delivery, the platelet count should be monitored and corticosteroids tapered slowly to avoid a rapid fall in platelet count and to ensure that the mother's mental state is not affected (evidence level IV).
Although prednisone is metabolized in the placenta by 11-beta-hydroxlase, high doses may have an effect on the fetus. A randomized study revealed no beneficial effect of low-dose maternal corticosteroids (1.5 mg of betamethasone orally per day) on the fetal platelet count152 (evidence level Ib).
IVIg.
If corticosteroid therapy is ineffective, significant side effects occur, or a more rapid platelet increase is required, IVIg should be considered. There are no comparative trials of corticosteroids and IVIg in pregnant women; however, response rates are similar to those in non-pregnant patients. After an initial response, single IVIg infusions are well tolerated and may be repeated as needed to prevent hemorrhage and provide an adequate platelet count for delivery (evidence level IV).
IV anti-D.
In non-splenectomized Rh(D)-positive patients, IV anti-D 50 to 75 μg/kg has been shown to be effective and safe for both mother and fetus in the second and third trimesters147,148 (evidence level IIb). In one study of anti-D for delivery, augmentation with corticosteroids or IVIg was usually required to achieve the required platelet count of 50 × 109/L.147 Although complications are uncommon, monitoring is required for neonatal jaundice, anemia, and direct antiglobulin test positivity after delivery.
Management options for maternal ITP patients failing first-line treatment.
Combining first-line therapy.
As with non-pregnant adults, combining first-line treatments in the refractory patient may be appropriate in the weeks before delivery (evidence level IV). HDMP (1000 mg) possibly in combination with IVIg or azathioprine has been suggested as a treatment for pregnant patients refractory to oral corticosteroids or IVIg143 (evidence level III). Data available for azathioprine in SLE and renal transplantation show that it is safe during pregnancy149-151 (evidence level III) but response is slow. Cyclosporin A has not been associated with significant toxicity to mother or fetus during pregnancy.153,154
If splenectomy is necessary, it is best performed in the second trimester and may be performed laparoscopically (evidence level III), but the technique may be difficult beyond 20 weeks' gestation. Appropriate vaccination during or after pregnancy is required.
Prepregnancy counseling.
It is rarely necessary to advise ITP patients against pregnancy. Counseling of ITP patients considering pregnancy should address safety of mother and fetus, outcomes of worsening maternal disease, and risks of pregnancy itself (evidence level IV).
Management of delivery.
Historically, management of delivery in mothers with ITP has been dominated by concerns over the risk of severe neonatal thrombocytopenia and hemorrhage (supplemental Document 8, Recommendation Box 10). In 1976, cesarean section was recommended for all ITP patients based on a reported perinatal mortality of 12% to 21%, largely resulting from birth trauma and ICH. However, these historical data were selective and excessively pessimistic. More recent reviews suggest the neonatal mortality rate of babies born to mothers with ITP is less than 1%.141 Large prospective studies published in the 1990s documented an incidence of “severe” neonatal thrombocytopenia (< 50 × 109/L) of 8.9% to 14.7%, with ICH occurring in 0% to 1.5% of infants with neonatal thrombocytopenia.141,155-157 There is no evidence that cesarean section is safer for the fetus with thrombocytopenia than uncomplicated vaginal delivery (which is usually safer for the mother). Moreover, most hemorrhagic events in neonates occur 24 to 48 hours after delivery at the nadir of the platelet count. Given the difficulty predicting severe thrombocytopenia in neonates and very low risk of serious hemorrhage (evidence level III, grade B recommendation), the mode of delivery in ITP patients should be determined by purely obstetric indications.143,158
Management of the neonate (of mothers with ITP).
Neonatal ITP (from mothers with ITP) accounts for only 3% of all cases of thrombocytopenia at delivery.137 In low-birth-weight neonates admitted to intensive care, thrombocytopenia develops in 18% to 35% of admissions and is most common in the smallest infants159 (evidence level IIb). Fetal or neonatal platelet count cannot be reliably predicted by maternal platelet count, platelet antibody levels, or history of maternal splenectomy for ITP141,143,160 (evidence level III). Fetal blood sampling by cordocentesis161 carries a fetal mortality risk of 1% to 2% (at least as high as the risk of ICH). Scalp blood sampling in early labor to measure fetal platelet count162 is technically difficult, can cause significant hemorrhage, and commonly produces misleading platelet count results because of clotting caused by exposure to vernix or amniotic fluid.163 Attempts to measure the fetal platelet count before delivery are not recommended (evidence level III). Procedures during labor associated with increased hemorrhagic risk to the fetus should be avoided (evidence level IV) including use of (1) fetal scalp electrodes, (2) fetal blood samples, (3) ventouse delivery, and (4) rotational forceps (evidence level IV).
After delivery, a cord blood platelet count should be determined157 (level III evidence, grade C recommendation) by clean venepuncture of a cord vessel rather than by draining blood from the cord. Intramuscular injections, such as vitamin K, should be avoided until the platelet count is known. Infants with subnormal counts should be observed clinically and hematologically, as platelet counts tend to nadir between days 2 and 5 after birth. Transcranial ultrasonography should be performed on neonates with platelet counts less than 50 × 109/L at delivery (evidence level IV). Although treatment of the neonate is rarely required, in those with clinical hemorrhage or platelet counts less than 20 × 109/L, treatment with a single dose of IVIg 1 g/kg (repeated if necessary) produces a rapid response. Life-threatening hemorrhage should be treated by platelet transfusion combined with IVIg. Severe thrombocytopenia and clinical hemorrhage in neonates are rare due to maternal ITP so when present, neonatal alloimmune thrombocytopenia should be excluded by laboratory testing.
Neonatal thrombocytopenia secondary to maternal ITP may last for months and requires long-term monitoring and occasionally a second dose of IVIg at 4 to 6 weeks after birth. In alloimmune thrombocytopenia, fetal thrombocytopenia tends to worsen in subsequent pregnancies.164 In ITP the second fetus is usually as affected as the first (evidence level III).
Obstetric analgesia and anesthesia.
The decision about regional anesthesia is ideally made before delivery in conjunction with the obstetric anesthetist (supplemental Document 8, Recommendation Box 11). The general trend in recent years has been to lower the “cutoff point” to 75 to 100 × 109/L.145 However, there are no data to support a minimum required platelet count and each case must be individually considered, with the risk of the procedure (spinal hematoma) balanced against benefits (pain relief, better blood pressure control, avoidance of general anesthesia). There are too few reports of epidural hematoma following regional blockade in obstetric patients to give an incidence of this complication.165
In the absence of bruising, bleeding history, and anticoagulation, and if the international normalized ratio (INR), activated partial thromboplastin time (APTT) test, and fibrinogen levels are normal, a small consensus of obstetric anesthetists agree no changes to routine practice are required until the platelet count drops below 50 × 109/L. For lower counts, a careful analysis of benefit against risk of epidural hematoma is needed, and multidisciplinary is discussion encouraged. Risk of vascular damage likely decreases proportionately to needle size, and consequently spinal may be a safer option than epidural blockade. An experienced operator is required (evidence level IV).
When monitoring platelet levels, the trend, as well as the absolute value, is important, and the mother with a rapidly falling count should be observed more closely than one with low but stable levels.166
Some maternity units have access to thromboelastography or other point-of-care global hemostasis monitoring.167 Using this technique, thrombocytopenia can be evaluated against the prothrombotic state of pregnancy, rather than monitoring platelet function alone. However, utility of these monitors in obstetric hemostasis has never been validated167 (evidence level IV).
Venous thromboembolism.
Despite thrombocytopenia, ITP in pregnancy may be associated with a prothrombotic state, due to anticardiolipin antibody syndromes, or other factors more common in pregnancy, and VTE prophylaxis should be considered.
Diagnostic approach in children with suspected ITP
The diagnosis of ITP in children is one of exclusion (supplemental Document 8, Recommendation Box 12; Table 1). Children with newly diagnosed ITP and atypical features should be referred to, or discussed with, a hematologist experienced in the assessment and treatment of children with ITP. Children and their parents may benefit from contacts and literature that are available from ITP support groups (including those mentioned previously).
Differential diagnosis
Although presentation is generally acute, bruising and purpura may develop slowly over weeks or months, suggesting a chronic course. It is important to exclude other common disorders that may resemble ITP (Table 2).
If a decision is made to observe the child with presumed newly diagnosed ITP, even in typical cases, a complete blood count and blood smear should be repeated periodically to exclude the evolution of a serious bone marrow or other hematologic disorder until the diagnosis is clear or recovery has occurred.
Children with familial inherited thrombocytopenias have sometimes been misdiagnosed as having ITP.168,169 Inherited disorders should be suspected if thrombocytopenia has been present since early life, a positive family history for a similar disorder is elicited,170 or characteristic features are present.
Special diagnostic considerations in children.
Older children are more likely to have chronic disease171 (evidence level III). Other autoimmune diseases associated with thrombocytopenia, including SLE, CVID, and autoimmune lymphoproliferative syndrome (ALPS [although difficult to assess in some instances]) should be considered in cases with multiple autoimmune cytopenias, as should HIV infection in those with risk factors for this infection.
Bone marrow evaluation.
Bone marrow evaluation in children with newly diagnosed ITP is recommended only when abnormalities are present other than isolated thrombocytopenia in the blood count/smear, if systemic features (eg, bone pain) are apparent, or if the patient has an otherwise unexplained enlarged spleen. Bone marrow evaluation should at least be considered in cases who respond minimally or not at all to first-line therapies.172
Helicobacter pylori infection, ANA, APLA (including lupus anticoagulant [LAC]).
See “Helicobacter pylori testing” in “Diagnostic approach in patients with suspected ITP.”
Examination of patients with persistent/early refractory ITP.
For patients with an initial diagnosis of ITP and no improvement in platelet count after 3 to 6 months and who still require treatment, several evaluations are recommended (Table 7).
Recommended evaluations for children newly diagnosed with ITP and no improvement after 3 to 6 months
|
|
Refer also to corresponding topics under “Management of adult ITP,” and supplemental Document 6.
ANA indicates antinuclear antibody; APLA, antiphospholipid antibodies; ACA, anticardiolipin antibody; and LAC, lupus anticoagulant.
Management of ITP in childhood: general measures
Only 3% of children with ITP have clinically significant symptoms such as severe epistaxis or GI bleeding173-177 (evidence level IIb/III). Severe bleeding is more likely in children with platelet counts less than 10 × 109/L178 (evidence level III). The incidence of ICH in children with ITP is approximately 0.1% to 0.5%171,179,180 (evidence level III), and predicting with confidence which children will develop an ICH is not possible. Risk factors for ICH in children with severe thrombocytopenia include head trauma and concomitant use of medications that adversely affect platelet function. Caution should be exercised in the management of children with ITP and coexisting vasculitis or coagulopathies, as may be seen in cases with varicella-associated ITP. Current opinion favors consideration of multiple factors when deciding to treat or not to treat children with ITP, including bleeding symptoms, the platelet count, and psychosocial and lifestyle issues such as activity profiles.
Clinical classification
Classification of children with ITP by severity of bleeding is useful to guide management (supplemental Document 8, Recommendation Box 13; Table 8).
Grade of severity and management of patients with ITP
| Bleeding/quality of life | Management approach |
|---|---|
| Grade 1. Minor bleeding, few petechiae (≤ 100 total) and/or ≤ 5 small bruises (≤ 3-cm diameter); no mucosal bleeding | Consent for observation |
| Grade 2. Mild bleeding, many petechiae (> 100 total) and/or > 5 large bruises (> 3-cm diameter); no mucosal bleeding | Consent for observation or for treatment in selected children |
| Grade 3. Moderate bleeding, overt mucosal bleeding, troublesome lifestyle | Intervention to reach grade 1/2 in selected children |
| Grade 4. Mucosal bleeding or suspected internal hemorrhage | Intervention |
| Bleeding/quality of life | Management approach |
|---|---|
| Grade 1. Minor bleeding, few petechiae (≤ 100 total) and/or ≤ 5 small bruises (≤ 3-cm diameter); no mucosal bleeding | Consent for observation |
| Grade 2. Mild bleeding, many petechiae (> 100 total) and/or > 5 large bruises (> 3-cm diameter); no mucosal bleeding | Consent for observation or for treatment in selected children |
| Grade 3. Moderate bleeding, overt mucosal bleeding, troublesome lifestyle | Intervention to reach grade 1/2 in selected children |
| Grade 4. Mucosal bleeding or suspected internal hemorrhage | Intervention |
Three bleeding scores confirm that most children with ITP do not have serious bleeding problems despite very low platelet counts.173,181,182 Of note, the severity of mucocutaneous bleeding does not predict the risk for life-threatening bleeding (eg, ICH), and children should not be treated based on cutaneous signs alone without consideration of other factors including the circulating platelet count, activity profile, and psychosocial issues.
Health-related quality of life in children
Patient-reported outcomes, including health-related quality of life (HRQoL) measures, are useful components for evaluating and understanding the effects of symptoms and treatments from the patient's perspective. A disease-specific tool has now been developed—the Kid's ITP Tools (KIT)—that has reliable management properties183 (evidence level IIa).
Expectant “watch and wait” policy
The majority of children with newly diagnosed ITP lack significant bleeding symptoms and may be managed without therapy directed at raising the platelet count at the discretion of the hematologist and the patient173-175,182,184-186 (evidence levels II-IV; supplemental Document 8, Recommendation Box 14). It is essential, therefore, that parents and children with ITP understand the risks of serious or life-threatening hemorrhage, and are also aware that children for whom drug therapy is prescribed are those at substantial risk of serious hemorrhage.
Hospitalization
For children with an established diagnosis of ITP, hospital admission should be reserved for those who have clinically significant bleeding. Problematic psychosocial circumstances of child and family (eg, behavioral issues, residence remote from a health care facility) should also be considered. Parents should be advised to watch for other signs of bleeding and be given a contact name and telephone number where a physician can be reached at all times. The child should not participate in competitive contact activities that have a high risk of head trauma. Other activities need not be restricted and the child should be encouraged to continue schooling (evidence level IV, grade C recommendation [supplemental Document 8, Recommendation Box 15]).
Most children with minor, mild, or moderate symptoms can be safely managed as outpatients with judicious use of supportive care (eg, antifibrinolytic agents, oral contraceptives) and weekly or less-frequent outpatient visits. When severe thrombocytopenia persists, limiting activities may be an option or treatment can be initiated. During teenage years, the issues of lifestyle and self-image assume greater importance and should also be discussed and may influence treatment decisions.
General measures for persistent and chronic ITP in children
The management of children with persistent/refractory ITP is essentially the same as those with newly diagnosed ITP. Many children stabilize with an adequate platelet count (> 20-30 × 109/L) and have no symptoms unless injured. Spontaneous remission may occur over time and expectant management can continue depending on the risk of bleeding and the degree of activity restriction of the child. The onset of menstruation may be problematic and can be managed with antifibrinolytic agents and hormonal medication (see “Supportive care” under “Management of adult ITP”). Children and parents should not forget the vulnerability to excessive bleeding following serious accidents and it is advisable for the family to carry a card or letter with details of the disorder in case of emergency. A medical bracelet or pendant may be appropriate (evidence level IV).
Some children with ITP will have platelet counts of 10 to 30 × 109/L and, although they have no serious bleeding, they are nonetheless troubled by purpura. Treatment may be beneficial in these cases. As adolescents, minors may become very conscious of their appearance and need sympathetic support.
Management of ITP in children
It is necessary to treat all children with severe bleeding symptoms and treatment should also be considered in children with moderate bleeding or those at increased risk of bleeding (Table 9).
First-line/initial treatment in children with ITP
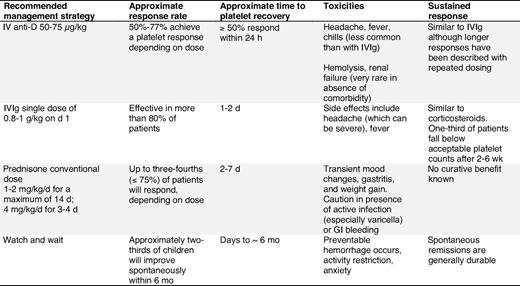
TPO-receptor agonists: studies in children are ongoing. At this time, evidence to support use of these agents in children is not available, but encouraging results have been reported in adults (see “Management of adult ITP”).
More complete details regarding the studies found in the literature search (2001-2008) are available in supplemental Document 7.
First-line/initial treatment options to raise platelet counts in children
See supplemental Document 8, Recommendation Box 16.
Intravenous anti-D immunoglobulin.
IV anti-D immunoglobulin can be given to Rh(D)-positive children as a short infusion and is usually effective in transiently raising platelet counts.47,48,52,187,188 Mild extravascular hemolysis is common and a few instances of intravascular hemolysis, disseminated intravascular coagulation, and renal failure have been reported in pediatric patients with comorbidities.44,53,189,190
Intravenous immunoglobulin (IVIg).
Predniso(lo)ne/corticosteroids.
Prednisone at a dose of 1 to 2 mg/kg/d may be effective at inducing a response in children15,16 (evidence level Ib). Higher doses (4 mg/kg/d) for 3 to 4 days have been shown to be effective in up to 72% to 88% of children (platelet count > 50 × 109/L) within 72 hours187,195,197 (evidence level Ib/III).
Because of the serious side effects associated with prolonged corticosteroid treatment in children with ITP, prednisone should be used only to maintain a hemostatic platelet count, and for as short a time as possible.
Emergency treatment in children
In organ- or life-threatening situations (as with adult patients), a larger-than-usual dose (2- to 3-fold) of platelets should be infused together with IV high-dose corticosteroids and IVIg or IV anti-D (supplemental Document 8, Recommendation Box 17). The goal of treatment is to elevate the platelet count to a level where the risk of severe bleeding is minimized as soon as possible. In special circumstances, emergency splenectomy may need to be considered (evidence level IV).
Treatment options for children with persistent or chronic ITP
The goal of treatment for children with persistent or chronic ITP is to maintain a hemostatic platelet count with first-line therapies (eg, IVIg, IV anti-D, short-course corticosteroids) and to minimize the use of prolonged corticosteroid therapy (supplemental Document 8, Recommendation Box 18). Cytotoxic drugs should be used with extreme caution in children. All children with persistent or chronic ITP should have their case reviewed and managed by a hematologist experienced in the diagnosis and management of children with ITP
Table 10 contains a summary of the treatment options for children with chronic ITP in whom first-line treatments are not successful. Treatment options are listed alphabetically and do not imply preference.
Treatment options in children with persistent or chronic ITP
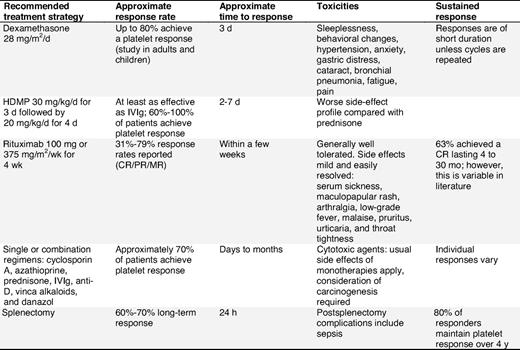
See supplemental Document 7 for relevant studies. More complete details regarding the studies found in the literature search (2001-2008) are available in the appendices.
HDMP indicates high-dose methylprednisolone; CR, complete response; PR, partial response; and MR, minor response.
Dexamethasone.
Dexamethasone (28-40 mg/m2/d) has been reported as treatment for patients with refractory ITP.198,199 The response rate in previously untreated patients aged 18 years or younger was 86%, with 67% of all evaluable patients reaching platelet levels of at least 50 × 109/L lasting for a median time of 26 months36 (evidence level IIa). However, side effects such as sleeplessness, aggressive behavior, and loss of concentration are unacceptably high.198
High-dose methylprednisolone.
HDMP (given as an oral 7-day course of 30 mg/kg/d for 3 days followed by 20 mg/kg/d for 4 days) has been used as an alternative to IVIg200-202 (evidence level Ib-III).
IVIg/anti-D.
See “First-line/initial treatment options to raise platelet counts in children.”
Rituximab (see “Rituximab” in “Second-line treatment options for adult ITP patients”).
Rituximab has been used with success in children with chronic refractory ITP203-205 (evidence levels IIa and IIb). Overall, the response rate (> 50 × 109/L platelet count) is between 31% and 68%. In all case series, rituximab was well tolerated with the exception of serum sickness (supplemental Document 7).
Single or combination regimens (see “Combination chemotherapy” in “Treatment options for adult patients failing first- and second-line therapies”).
The experience in children is limited and therefore no recommendations can be made65 (evidence level IIb/IV).
TPO-receptor agonists.
A number of studies with TPO-receptor agonists have shown encouraging results in adults.90,92-98 However, no finalized studies in children are available to support the use of these agents in this patient population. Assuming that the long-term safety of these agents is confirmed, they could be used not only for children with chronic refractory ITP, but also in those with persistent but highly symptomatic disease resistant to usual first-line treatments.
Surgical options for children with chronic refractory ITP
Bleeding complications of ITP must be managed according to severity and circumstances, and splenectomy is generally deferred for as long as possible. Seventy to 80% of children will initially respond to splenectomy but the postoperative risk of infection is a deterrent to its routine use.
Splenectomy.
Splenectomy with appropriate previous vaccination, followed by prophylactic antibiotics, is an effective treatment for pediatric ITP (supplemental Document 8, Recommendation Box 19). However, it is rarely recommended in children because the risk of death from ITP in childhood is extremely low (< 0.5%). The comparative figure associated with postsplenectomy overwhelming sepsis is up to 3% in children.206 The risk of sepsis probably persists for life. In children who do undergo splenectomy, the overall effectiveness is good, but complications, primarily sepsis, remain a concern206,207 (evidence level IIb/III).
Therapies whose use is no longer justified
Consensus regarding a lack of efficacy or high reported toxicity, or because of a lack of evidence means that interferon-α and campath-1H can no longer be justified for use in childhood ITP (evidence level IV).
Conclusions
Despite significant progress in our understanding of the pathophysiology and management of ITP during the past 15 years, surprisingly few RCTs have been conducted and evidence-based guidelines to inform management of individual patients are limited. There are also few validated risk factors regarding outcome prediction or response to therapies (including splenectomy) and little progress has been made toward making ITP anything other than a “diagnosis of exclusion.” Consequently, future research on ITP must be focused on carefully designed randomized trials and multicenter prospective cohort studies.
Addendum
Anti-D immunoglobulin may not be available in all countries, and in June 2009, the European Medicines Agency was formally notified by Cangene Europe Ltd of its decision to withdraw all its applications and all marketing authorizations for their anti-D immunoglobulin product WinRho SDF.
The online version of this article contains a data supplement.
Acknowledgments
The writing and project management of this consensus document were supported by an unrestricted grant provided by Amgen Ltd, Baxter Ltd, and GlaxoSmithKline Ltd. Frances Essex, MSc, Senior Medical Writer, ScopeMedical Ltd, provided writing assistance throughout the project; Rebecca Hunt, Account Director, ScopeMedical Ltd, provided project management; and Keely Jennings BA, Editorial Services Manager, ScopeMedical Ltd, provided editorial assistance throughout the project.
Authorship
Contribution: D.P., A.C.N., D.J.K., R.S., D.B.C., P.A.I., V.S.B., B.J.H., and T.B.G. wrote the paper; D.P., D.J.K., R.S., and V.S.B. analyzed data; A.C.N. led the manuscript review; D.J.K. led manuscript review for adult ITP; R.S. and D.B.C. led manuscript review for the diagnosis of ITP; P.A.I. and V.S.B. led manuscript review for pediatric ITP; B.J.H. and T.B.G. led manuscript review for ITP in pregnancy; and J.G., P.B-M., M.T., I.G., G.L., R.M., J.B.B., F.R., B.H.C, B.G., M.A.S., S.W., and J.Y. reviewed the manuscript.
Conflict-of-interest disclosure: D.P. serves as a consultant for Amgen, GlaxoSmithKline (GSK), Shionogi, ONO, and Eisai; receives research support from Amgen, GSK, and Baxter Healthcare; and serves on the speakers bureau for Amgen. R.S. receives honoraria for participation on advisory boards and/or as a speaker at medical education events supported by GSK and Amgen. A.C.N. receives research funding from Amgen, Baxter, GSK, and Genentech; serves on speaking panels for Amgen and GSK and on advisory boards for GSK and PanGenetics; has no financial interest (stocks and shares); is not employed by any relevant company; and has no patents pending. V.S.B. serves on the Bayer International Haemophilia Advisory Board and is a member and chair of Novo Nordisk PRO-PACT Advisory Board. P.B.M. receives support from Baxter for travel to international meetings (International Society on Thrombosis and Haemostasis Scientific and Standardization Committee meeting, Vienna, Austria, 2008, and American Society of Hematology meeting, New Orleans, LA, 2009); participates in advisory board meetings for both Amgen and GSK; and served as chair of a satellite meeting for Baxter at the 2009 British Society for Haematology meeting. J.B.B. receives clinical research support from Amgen, Biogen-IDEC, Cangene, Genentech, GSK, Genzyme, Immunomedics, Ligand, MGI Pharma/Eisai Inc, and Sysmex; participates in speakers bureau programs for Baxter, Amgen, and GSK; owns stock in Amgen and GSK; participates in advisory boards for Amgen, GSK, Ligand, and Baxter; and participates in discussions of unlabeled or investigational use of product(s) that might include anti-cd40 ligand, thrombopoietic agents (AMG531 [Amgen], Eltrombopag [GSK], AKR501 [AkaRx]), Rigel, and rituximab [Genentech/Biogen-IDEC]. B.H.C. is a member of the scientific advisory boards of GSK, Amgen, Commonwealth Serum Laboratory (CSL), Bayer, and Boehringer Ingelheim; and serves as a consultant for CSL and Boehringer Ingelheim. D.B.C. serves as a consultant for Amgen, GSK, Symphogen, Genzyme, and Shionogi. T.B.G. receives research funding from and serves as a consultant for Amgen and GSK. B.G. serves as a consultant for Roche France, Amgen, and Laboratoire de fractionnement et de Biotechnologies (LFB; Les Ulis, France). J.G. receives honoraria for participation on advisory boards for GSK, Amgen, and Baxter, and receives research support from Baxter. B.J.H. lectures at meetings sponsored by Baxter. P.A.I. receives research funding from Amgen, GSK, and Roche, and serves as a consultant for Symphogen. R.M. owns stock in GSK and serves as ad hoc consultant for Amgen and GSK. F.R. receives honoraria for participation in advisory boards and/or as a speaker at medical education events supported by Amgen, GSK, and Shionogi. D.J.K. receives research support from and serves as a consultant for GSK, Amgen, MGI Pharma, Ligand, ONO, and Shionogi. I.G., G.L., M.A.S., M.T., S.W., and J.Y. declare no competing financial interests.
Correspondence: Drew Provan, Barts and The London School of Medicine and Dentistry, Dept of Haematology, 80 Newark St, London, E1 2ES, United Kingdom; e-mail: a.b.provan@qmul.ac.uk.
