

Abstract
Despite recent advances in our understanding of biochemical regulation of neutrophil chemotaxis, little is known about how mechanical factors control neutrophils' persistent polarity and rapid motility. Here, using a human neutrophil-like cell line and human primary neutrophils, we describe a dynamic spatiotemporal pattern of tractions during chemotaxis. Tractions are located at both the leading and the trailing edge of neutrophils, where they oscillate with a defined periodicity. Interestingly, traction oscillations at the leading and the trailing edge are out of phase with the tractions at the front leading those at the back, suggesting a temporal mechanism that coordinates leading edge and trailing edge activities. The magnitude and periodicity of tractions depend on the activity of nonmuscle myosin IIA. Specifically, traction development at the leading edge requires myosin light chain kinase-mediated myosin II contractility and is necessary for α5β1-integrin activation and leading edge adhesion. Localized myosin II activation induced by spatially activated small GTPase Rho, and its downstream kinase p160-ROCK, as previously reported, leads to contraction of actin-myosin II complexes at the trailing edge, causing it to de-adhere. Our data identify a key biomechanical mechanism for persistent cell polarity and motility.
Introduction
Chemotaxis, the directed movement of cells in a gradient of chemoattractant, is essential for neutrophils to crawl to sites of inflammation and infection. Chemoattractant-induced activation of spatially localized cellular signals causes neutrophils to initiate polymerization of actin at the leading edge (pseudopod), polarize (ie, adopt an asymmetric shape with defined front and back), and move toward the highest concentration of the chemoattractant. Recent studies have begun to reveal some fascinating details of the intracellular biochemical components that spatially direct the neutrophils' cytoskeleton and the complex signaling pathways that control formation of their front and back.1-4 Divergent frontness and backness signals provide a mechanism for neutrophils to polarize in uniform concentrations of chemoattractant and to perform U-turns rather than simply reverse polarity in response to changes in the direction of the attractant gradient.4
Despite these findings, there are significant gaps in our understanding of the mechanical functions that control the persistent and rapid movement of neutrophils. Specifically, the spatial and temporal dynamics, regulation, and functions of tractions remain largely undefined in neutrophils. It is well established that slow-moving cells, such as fibroblasts, assemble transient adhesions called focal complexes at the leading edge, which mature into more stable focal adhesions.5 Focal adhesions provide robust anchors to the extracellular matrix (ECM), allowing actomyosin-based stress fibers to pull the cell body forward. Tractions are transmitted to the substrate at the site of focal adhesions and are required for maturation of these adhesion structures.6 In contrast, focal adhesions and stress fibers are not detected in migratory neutrophils or T cells,4,7 raising the question whether and how mechanical forces control adhesion and directional migration in these rapidly moving amoeboid cells.
In this study, we revealed a highly dynamic spatiotemporal pattern of tractions in neutrophils during chemotaxis. The pattern is conserved in a human neutrophil-like cell line and primary human neutrophils and depends on nonmuscle myosin IIA. We show that spatiotemporal organization of tractions requires localization-specific myosin II activation and is essential for leading edge adhesion and trailing edge de-adhesion. These data reveal a biomechanical mechanism that promotes the rapid and highly coordinated movements in neutrophils during chemotaxis.
Methods
Cell culture and transfection
Cultivation and differentiation of HL-60 cells were as described.2 For transient transfections, the AMAXA nucleofection system was used. Differentiated HL-60 cells (2 × 107, on days 5-6 after dimethyl sulfoxide addition) were spun down and resuspended in nucleofector solution V. DNA (5 μg) or siRNA (3 μg) was added to the cells, and the cell-DNA mixture was subjected to nucleofection (program T-19). Nucleofected cells were transferred to 20 mL of complete medium. Subsequent assays were performed 3 to 6 hours for the expression vectors and 24 to 48 hours for siRNAs after transfection.
Isolation of primary neutrophils
Primary neutrophils were isolated from venous blood from healthy human donors. Blood was collected into heparin-containing Vacutainer tubes (BD Biosciences), and neutrophil isolation procedure was performed within 30 minutes of blood collection using polymorphonuclear leukocyte isolation medium (Matrix). Red blood cell contaminants were removed by Red Blood Cell Lysis buffer (Roche Diagnostics), which produced more than 97% of neutrophil purity. Neutrophils were suspended in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37°C until the time of experiments conducted within 8 hours after isolation.
Immunofluorescence and live-cell imaging
For immunofluorescence in fixed cells, cells were stimulated with 1μM formyl-methionyl-leucyl-phenylalanine (fMLP) in modified HBSS buffer for the indicated time. Cells were extracted with 0.2% Triton X-100 for 10 minutes at room temperature, fixed in 3.7% paraformaldehyde, and immunostained. Antibodies were used at dilutions of 1:50 (myosin light chain kinase [MLCK]), 1:200 [myosin regulatory light chain phosphorylated at Ser19 (p[19]-MRLC)], 1:500 (myosin heavy chain IIA), and 1:250 (α5-integrin), and immunostaining was performed as described.2 F-actin was assessed by incubation of cells with 0.2 unit of AlexaFluor-488–conjugated phalloidin (10 minutes at room temperature).
For live-cell imaging, cells were plated on human fibronectin (FN)–coated surface and stimulated either with a uniform concentration of 1μM fMLP or a point source of 10μM fMLP from a micropipette (internal diameter of 1 μm, pulled from a glass capillary), as described.8 Differential interference contrast (DIC) images, fluorescent images, and combined DIC/fluorescence images were collected with a Carl Zeiss 40×/1.30 NA Fluar DIC objective or a 63×/1.4 NA Plan Apochromat DIC objective on a Carl Zeiss Axiovert 200M microscope. All images were collected with a cooled charge-coupled device camera (AxioCam MR3, Carl Zeiss) and processed by ImageJ software 1.43.
For total internal reflection fluorescence (TIRF) microscopy, a TIRF module based on Axiovert 200M microscope (Carl Zeiss) was used as a starting point. Before each experiment, 488-nm excitation light from a 100-mW multiline argon-ion laser (Carl Zeiss) was aligned properly to create evanescent wave that can only excite the molecules within a layer of 100 nm above the cover glass. At the start of an experiment, a FN-coated cover glass to which cells were attached was placed on α Plan-Fluar 100×/1.45 oil objective lens. Microscopic images from the TIRF-illuminated cells were captured by AxioCam MR3 camera with an interval of 5 seconds. Exposure times for each image were typically less than 1 second, and the TIRF laser was shuttered with a Uniblitz COM2 shutter between exposures to minimize photodamage and photobleaching effects.
Polyacrylamide gel substrates, TFM, and data analysis
Polyacrylamide gels were prepared as described.9 Red fluorescent microspheres (0.2 μm, Invitrogen) were embedded in gels for traction detection, and the gels were coated with 10 μg/mL human FN. The elastic Young modulus of the polyacrylamide gels was 3.5 kPa (5% acrylamide; 0.10% bisacrylamide)10,11 ; 100-kPa gels were generated as described by Oakes et al.12 Briefly, differentiated HL-60 (dHL-60) cells or primary neutrophils (1.0 × 105) with or without various treatments were allowed to adhere to the FN-coated polyacrylamide gel for 10 minutes and migrate toward the micropipette containing 10μM fMLP. Traction force microscopy (TFM) was performed using Andor Technology Revolution System Spinning Disk Confocal Microscopy system (Andor) coupled to an Olympus IX71 inverted microscope with Olympus 100×/1.4 APO NA objective. The working distance of the objective is approximately 0.13 mm. HL-60 cells stably expressing green fluorescent protein (GFP) were used for TFM to track the outline of cells during the migration. Primary neutrophils were labeled with calcein AM (30 minutes) or DiO (5 minutes) and washed once with RPMI 1640 media before imaging studies. The 532-nm (20 mW) and 635-nm (25 mW) diode-pumped solid state laser lines were used to excite the fluorophores within the cell and the beads. The exposure time for both channels was 21 msec. Using iXon EM + DU-897 back illuminated EMCCD (Andor Technology), images of cells expressing GFP and fluorescent beads were captured interchangeably every 0.8 seconds for each fluorescent channel during stimulation and after cell detachment from the substrate. Images of the beads were analyzed by a custom-made program to calculate bead displacement and to generate traction maps. Images of beads after cell detachment, therefore traction-free, were used as a reference.
For traction analysis, the leading edge of a polarized neutrophil was defined as the area within the first 3 μm of the cell (for control cells and cells treated with Y-27632), whereas the rest of the cell was defined as the trailing edge. For ML-7–treated, MLCK-depleted or blebbistatin-treated cells, the leading edge was defined as the area within the first 2.2 μm of the cell. These definitions were based on quantifications of the leading-edge areas in HL-60 cells expressing the frontness markers, such as YFP-actin, and primary neutrophils stained with fluorescent phalloidin (for F-actin) with or without the drug treatments or MLCK depletion. The average tractions at the leading and trailing edges were calculated from the traction maps generated by the use of TFM.
To determine the periodicity of the front and rear tractions, Fourier analysis was performed using the traction values obtained from the TFM. The analysis was done using the built-in MATLAB “fft” (Fast Fourier Transform [FFT]) function to transform the original time-domain traction signals into frequency domain. The power spectral density (PSD) plots were then generated based on the results from the Fourier transform analysis.
To analyze cross-correlation between tractions at the leading and trailing edges and to calculate the cross-correlation coefficient (r),13 a customized MATLAB (Mathworks) program was developed:
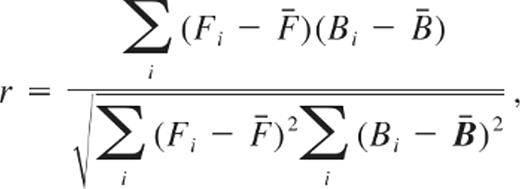
where Fi and Bi are the mean traction values of the front and back at time point i, and F̄ and B̄ are the averaged mean traction values of the front and back over entire time. To examine the time lag between tractions at the leading and trailing edges, the values for tractions at the front were shifted in time with those at the back fixed.
Integrin activation assay
A GST-tagged protein composed of ninth, 10th, and 11th FN type III repeats (denoted as GST-FN III9-11) was used to monitor active α5β1-integrin as reported.14 The GST-FN III9-11 fragment binds to unoccupied active integrins only. For adherent cells, integrin activation was determined as follows. After an exposure to chemoattractant or buffer, cells were fixed with paraformaldehyde (3.7%) and incubated with 50 μg/mL GST-FN III9-11 in phosphate-buffered saline with 1mM Ca2+ and 1mM Mg2+ at 37°C for 30 minutes. Because the GST-FN III9-11 fragment competes with the FN substrate to an extent, fixation of cells could eliminate interference from the FN substrate. After incubation with GST-FN III9-11, cells were gently washed, lysed in sodium dodecyl sulfate sample buffer, and bound GST-FN III9-11 was blotted with an anti-GST antibody (1:1000). For immunofluorescence studies, after fixation cells were incubated with GST-FN III9-11, followed by incubation with GST antibody and Alexa-conjugated secondary antibody. For suspended cells, a similar procedure was used, except that the cells were not fixed. A rabbit anti–α5-integrin antibody (1:1000) was used to assess the total level and localization of α5-integrin.
Methods for other experimental procedures are available in the supplemental data (available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Results
Spatial and temporal organization of tractions during neutrophil chemotaxis
To assess the spatial and temporal pattern of tractions in neutrophils during chemotaxis, we plated cells on a flexible substrate coated with FN and stimulated them with a point source of the chemoattractant fMLP. Fibronectin has been widely used as a substrate for the study of neutrophil chemotaxis.2,12,15 We used a substrate stiffness of 3.5 kPa, resembling the stiffness of tissue cells such as endothelial cells (1-5 kPa).16 We used both the neutrophil-like dHL-60 cells and human primary neutrophils. HL-60 cells, when differentiated, exhibit neutrophil-like morphologies, polarize and migrate in gradients (or in uniform concentrations) of attractants at rates comparable with those of neutrophils from peripheral blood, but unlike primary neutrophils they can be continuously cultured and are genetically tractable.2,4,8 Therefore, HL-60 cells have been used widely as a model for studying neutrophil polarity and chemotaxis.17-20 We detected tractions at both the leading and the trailing edge of dHL-60 cells (Figure 1A). Tractions were not distributed evenly at the leading or the trailing edge but instead were often concentrated in small regions that ranged from 1 to 4 μm in diameter (Figure 1A; supplemental Video 1). Time-course analysis of tractions in cells moving toward fMLP revealed a highly dynamic temporal pattern: the mean level of tractions at both edges oscillated during migration (100-400 Pa; Figure 1A-B; supplemental Figure 1A). The average tractions are summarized in Table 1. FFT analysis of the time-domain signals revealed periodicity of tractions in chemotaxing dHL-60 cells: the PSD plots of results from FFT analysis demonstrated a single pronounced peak at 4.8 plus or minus 1.4 seconds for tractions at the leading edge and 4.8 plus or minus 1.6 seconds for those at the trailing edge (mean ± SEM, n = 9 cells; Figure 1C; supplemental Figure 1B). Primary human neutrophils showed a similar pattern of tractions. Tractions were detected at both edges, where they oscillated at a periodicity of 4.7 plus or minus 1.5 seconds and 4.7 plus or minus 1.2 seconds, respectively (mean ± SEM, n = 6 cells; Figure 1D). Interestingly, oscillations of tractions at the 2 edges were out of phase (Figure 1A; supplemental Figure 1A), and the tractions at the front preceded those at the back by 0.80 plus or minus 0.23 seconds (mean ± SEM, n = 9 cells; P < .0001) for dHL-60 cells and 0.40 plus or minus 0.16 seconds (mean ± SEM, n = 6 cells; P < .001) for primary cells, as shown by a cross-correlation analysis, which computed the time offsets between tractions at the front and the back by shifting the 2 datasets relative to each other (Figure 1E-F). The conserved periodic and out-of-phase behavior of tractions at the neutrophil's front and back suggests a biomechanical mechanism that drives coordinated movements of neutrophils via differential contractile stresses exerted onto the substrate.

Spatiotemporal dynamics of tractions during neutrophil chemotaxis. (A) dHL-60 cells were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated elastic polyacrylamide gel for the times indicated. The speed for cell migration is approximately 2.0 μm/minute, lower than the speed of cells on FN-coated glass (∼ 2.4 μm/minute). The cells also exhibit normal tail retraction on the elastic gel (data not shown). Traction maps of the cell are shown. Pseudocolor bar representing tractions is given in Pascal (Pa). Scale bar represents 5 μm. Arrow indicates the direction of fMLP gradient. The leading edge of a polarized neutrophil was defined as the area within the first 3 μm of the cell (marked by a white line), whereas the rest of the cell was defined as the trailing edge (“Polyacrylamide gel substrates, TFM, and data analysis”). The image series shows part (7.2 seconds, for which the cell traveled ∼ 0.24 μm) of the whole migratory response. The video of the cell in panel A is available in supplemental data. (B) Time series of traction maps from panel A (with 3 additional time points) was analyzed by a customized MATLAB program to determine the average tractions in both leading edge (front) and trailing edge (back) of the cells in a time-dependent manner. The graph shows part (∼ 9 seconds) of the whole migratory response. The x-axis indicates time in seconds; y-axis is in Pascal (Pa). The mean levels of tractions at the leading and the trailing edges were comparable. A graph of another cell with a longer migratory response is shown in supplemental Figure 1A. (C) PSD plots of tractions at the leading (left panel) and the trailing edge (right panel) of a migratory dHL-60 cell. PSD plots were generated based on the results from Fourier analysis of the traction values. The y-axis represents the power spectral density normalized to the highest peak value (= 1); x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Nine cells were analyzed, and a representative cell is shown. PSD plots of tractions in 3 cells combined are shown in supplemental Figure 1B. (D) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory primary neutrophil. Six cells were analyzed, and data from a representative cell are shown. (E) Left panel: Cross-correlation between tractions at the leading edge and the trailing edge against time offset during migration for individual dHL-60 cells. Dotted lines indicate zero offset. Note that the back traction lags the front traction as indicated by the maximum cross-correlation at time offset of 0.8 seconds. Data from 3 representative cells are shown. Time bar represents 24 seconds. Right panel: Summary of time offsets between leading edges and trailing edges (n = 9 cells) in dHL-60 cells. (F) Left panel: Cross-correlation between tractions at the leading and the trailing edges against time offset during migration for individual primary neutrophils. Data from 2 representative cells are shown. Right panel: Summary of time offsets between leading edges and trailing edges (n = 6 cells) in primary neutrophils.
Spatiotemporal dynamics of tractions during neutrophil chemotaxis. (A) dHL-60 cells were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated elastic polyacrylamide gel for the times indicated. The speed for cell migration is approximately 2.0 μm/minute, lower than the speed of cells on FN-coated glass (∼ 2.4 μm/minute). The cells also exhibit normal tail retraction on the elastic gel (data not shown). Traction maps of the cell are shown. Pseudocolor bar representing tractions is given in Pascal (Pa). Scale bar represents 5 μm. Arrow indicates the direction of fMLP gradient. The leading edge of a polarized neutrophil was defined as the area within the first 3 μm of the cell (marked by a white line), whereas the rest of the cell was defined as the trailing edge (“Polyacrylamide gel substrates, TFM, and data analysis”). The image series shows part (7.2 seconds, for which the cell traveled ∼ 0.24 μm) of the whole migratory response. The video of the cell in panel A is available in supplemental data. (B) Time series of traction maps from panel A (with 3 additional time points) was analyzed by a customized MATLAB program to determine the average tractions in both leading edge (front) and trailing edge (back) of the cells in a time-dependent manner. The graph shows part (∼ 9 seconds) of the whole migratory response. The x-axis indicates time in seconds; y-axis is in Pascal (Pa). The mean levels of tractions at the leading and the trailing edges were comparable. A graph of another cell with a longer migratory response is shown in supplemental Figure 1A. (C) PSD plots of tractions at the leading (left panel) and the trailing edge (right panel) of a migratory dHL-60 cell. PSD plots were generated based on the results from Fourier analysis of the traction values. The y-axis represents the power spectral density normalized to the highest peak value (= 1); x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Nine cells were analyzed, and a representative cell is shown. PSD plots of tractions in 3 cells combined are shown in supplemental Figure 1B. (D) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory primary neutrophil. Six cells were analyzed, and data from a representative cell are shown. (E) Left panel: Cross-correlation between tractions at the leading edge and the trailing edge against time offset during migration for individual dHL-60 cells. Dotted lines indicate zero offset. Note that the back traction lags the front traction as indicated by the maximum cross-correlation at time offset of 0.8 seconds. Data from 3 representative cells are shown. Time bar represents 24 seconds. Right panel: Summary of time offsets between leading edges and trailing edges (n = 9 cells) in dHL-60 cells. (F) Left panel: Cross-correlation between tractions at the leading and the trailing edges against time offset during migration for individual primary neutrophils. Data from 2 representative cells are shown. Right panel: Summary of time offsets between leading edges and trailing edges (n = 6 cells) in primary neutrophils.
Magnitude of tractions in migrating neutrophils
| Front, Pa | Back, Pa | |
|---|---|---|
| dHL-60 cells, 3.5 kPa | ||
| Control | 180 ± 39 | 207 ± 27 |
| ML-7 | 111 ± 26 | 118 ± 30 |
| MLCK KD | 110 ± 16 | 145 ± 54 |
| Blebbistatin | 136 ± 19 | 112 ± 14 |
| Y-27632 | 99 ± 16 | 87 ± 18 |
| Primary neutrophils, 3.5 kPa | ||
| Control | 56 ± 17 | 85 ± 30 |
| ML-7 | 40 ± 5 | 55 ± 7 |
| Blebbistatin | 33 ± 6 | 56 ± 11 |
| Y-27632 | 33 ± 8 | 27 ± 3 |
| Primary neutrophils, 100 kPa | ||
| Control | 596 ± 93 | 638 ± 102 |
| ML-7 | 399 ± 87 | 375 ± 37 |
| Blebbistatin | 310 ± 88 | 390 ± 67 |
| Y-27632 | 361 ± 83 | 349 ± 49 |
| Front, Pa | Back, Pa | |
|---|---|---|
| dHL-60 cells, 3.5 kPa | ||
| Control | 180 ± 39 | 207 ± 27 |
| ML-7 | 111 ± 26 | 118 ± 30 |
| MLCK KD | 110 ± 16 | 145 ± 54 |
| Blebbistatin | 136 ± 19 | 112 ± 14 |
| Y-27632 | 99 ± 16 | 87 ± 18 |
| Primary neutrophils, 3.5 kPa | ||
| Control | 56 ± 17 | 85 ± 30 |
| ML-7 | 40 ± 5 | 55 ± 7 |
| Blebbistatin | 33 ± 6 | 56 ± 11 |
| Y-27632 | 33 ± 8 | 27 ± 3 |
| Primary neutrophils, 100 kPa | ||
| Control | 596 ± 93 | 638 ± 102 |
| ML-7 | 399 ± 87 | 375 ± 37 |
| Blebbistatin | 310 ± 88 | 390 ± 67 |
| Y-27632 | 361 ± 83 | 349 ± 49 |
Myosin II controls tractions and is required for neutrophil chemotaxis
Myosin II filaments can apply forces to move actin filaments relative to each other. During fibroblast migration, the contractile force generated by the actin-myosin cytoskeleton causes tractions on the underlying substrate by propagating through the actin-integrin-ECM link.21 We therefore assessed myosin II expression and spatial localization in neutrophils. Experiments with isoform-specific antibodies demonstrated that both dHL-60 cells and primary neutrophils mainly expressed myosin IIA (supplemental Figure 2A), consistent with an earlier report.22 We then assessed the spatial localization of myosin II using antibodies against total myosin IIA, myosin regulatory light chain (MRLC) phosphorylated at Ser19 (p[19]-MRLC,4 the activated form of MRLC), and an mCherry-tagged myosin IIA (mCherry–myosin IIA)23 in fixed or live dHL-60 cells. Both total and activated myosin II and the mCherry-tagged fusion protein were located mostly at the trailing edge of polarized neutrophils (69%, 68%, and 72% of total fluorescence intensity for myosin IIA, p[19]-MRLC, and mCherry–myosin IIA, respectively; Figure 2A-B; supplemental Videos 2-3), consistent with earlier reports.4,24 There was a considerable amount of myosin IIA localization at the front (31%, 32%, and 28%, respectively; Figure 2A-B), as described previously in studies in primary neutrophils.15,25 Using confocal fluorescence microscopy combined with 3-dimensional reconstruction, we found that myosin IIA was enriched around the periphery of the uropod (trailing edge) and distributed vertically and somewhat diffusely in the pseudopodia (supplemental Figure 2B; supplemental Videos 4-5). Interestingly, although F-actin was highly enriched at the leading edge, it was also detected throughout the periphery of the cell, including at the back of the cell (supplemental Figure 2B; supplemental Videos 4-5).
![Figure 2. Myosin II controls tractions and is necessary for neutrophil chemotaxis. (A) dHL-60 cells were stimulated for 3 minutes by a uniform concentration of 1μM fMLP. Cells were fixed with 3.7% paraformaldehyde and stained with a specific antimyosin heavy chain IIA (MHCIIA) antibody, anti-p[Ser19]MRLC antibody, and rhodamine-conjugated phalloidin to localize filamentous actin (F-actin). The corresponding DIC image is also shown. Scale bar represents 10 μm. (B) dHL-60 cells were transfected with mCherry–myosin IIA (mChe-myoIIA) and stimulated by a micropipette containing 10μM fMLP for the indicated times. mCherry–myosin IIA fluorescence and the corresponding DIC images are shown. Arrows point to the locations of myosin IIA at the leading and trailing edges. Scale bar represents 10 μm. The video of the cell in panel B is available in supplemental data. (C) dHL-60 cells pretreated with blebbistatin (100μM, 30 minutes) were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated elastic polyacrylamide gel for the indicated times. Cells treated with blebbistatin migrated at 1.0 μm/minute on the elastic gel. Traction maps of the cell are shown. Pseudocolor bar representing traction force is given in Pascal (Pa). Scale bar represents 10 μm. The leading edge (within the first 2.2 μm of the cell) is marked by a white line (“Polyacrylamide gel substrates, TFM, and data analysis”). The image series shows part (5.6 seconds) of the whole migratory response. (D) Time series of traction maps from panel C (with 4 additional time points) was analyzed by a customized MATLAB program to determine the average tractions in both leading edge (front) and trailing edge (back) of the cells in a time-dependent manner. The graph shows part (∼ 9.6 seconds) of the whole migratory response. The x-axis indicates time in seconds; y-axis is in Pascal (Pa). (E) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory cell pretreated with blebbistatin (100μM, 30 minutes). The whole migratory response was analyzed. The y-axis represents the power spectral density normalized to the highest peak value (1); x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Cells depleted of myosin IIA exhibited similar response (not shown). Ten cells were analyzed, and a representative cell is shown. Additional plots are shown in supplemental Figure 2C. (F) Before exposure to attractant supplied by a micropipette containing 10μM fMLP, cells were not pretreated (control), were pretreated with blebbistatin (Blebbis, 100μM, 30 minutes), were infected with lentivirus-containing myosin IIA–targeting shRNAs (MyoII KD), or were pretreated with Y-27632 (30μM, 30 minutes). The 3 images in each row show the positions of individual cells (each identified with a superimposed letter) after the indicated times of exposure to fMLP. White and black arrows point to the poorly developed leading edges and long stretched tails, respectively. Cells infected with virus containing a scramble shRNA exhibited similar response to uninfected control cells. Lower doses of blebbistatin (≤ 50μM) were tested, which required a prolonged period of incubation to exert the same effects as 100μM blebbistatin (data not shown). Scale bar represents 10 μm. Videos of cells with or without treatments are available in supplemental data. (G) DIC kymographs of a dHL-60 cell untreated or treated with inhibitors or myosin IIA shRNAs migrating toward an fMLP (10μM)–containing micropipette. The left panel shows a portion of the neutrophils' leading edge under various conditions. The dotted rectangles indicate the regions of the cell used to generate the kymographs (before fMLP stimulation). The actual lengths of the rectangles are 20 μm in the direction of the arrow. White scale bar represents 1 μm. The right panel shows the DIC kymographs. White scale bar represents 5 μm. In both panels, white arrows indicate the direction of protrusion. Cells a, d, f, and h in panel F were used for the analysis. Approximately 8 minutes of migration was recorded.](/view-large/figure/7113590/zh89991058490002.jpeg)
Myosin II controls tractions and is necessary for neutrophil chemotaxis. (A) dHL-60 cells were stimulated for 3 minutes by a uniform concentration of 1μM fMLP. Cells were fixed with 3.7% paraformaldehyde and stained with a specific antimyosin heavy chain IIA (MHCIIA) antibody, anti-p[Ser19]MRLC antibody, and rhodamine-conjugated phalloidin to localize filamentous actin (F-actin). The corresponding DIC image is also shown. Scale bar represents 10 μm. (B) dHL-60 cells were transfected with mCherry–myosin IIA (mChe-myoIIA) and stimulated by a micropipette containing 10μM fMLP for the indicated times. mCherry–myosin IIA fluorescence and the corresponding DIC images are shown. Arrows point to the locations of myosin IIA at the leading and trailing edges. Scale bar represents 10 μm. The video of the cell in panel B is available in supplemental data. (C) dHL-60 cells pretreated with blebbistatin (100μM, 30 minutes) were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated elastic polyacrylamide gel for the indicated times. Cells treated with blebbistatin migrated at 1.0 μm/minute on the elastic gel. Traction maps of the cell are shown. Pseudocolor bar representing traction force is given in Pascal (Pa). Scale bar represents 10 μm. The leading edge (within the first 2.2 μm of the cell) is marked by a white line (“Polyacrylamide gel substrates, TFM, and data analysis”). The image series shows part (5.6 seconds) of the whole migratory response. (D) Time series of traction maps from panel C (with 4 additional time points) was analyzed by a customized MATLAB program to determine the average tractions in both leading edge (front) and trailing edge (back) of the cells in a time-dependent manner. The graph shows part (∼ 9.6 seconds) of the whole migratory response. The x-axis indicates time in seconds; y-axis is in Pascal (Pa). (E) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory cell pretreated with blebbistatin (100μM, 30 minutes). The whole migratory response was analyzed. The y-axis represents the power spectral density normalized to the highest peak value (1); x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Cells depleted of myosin IIA exhibited similar response (not shown). Ten cells were analyzed, and a representative cell is shown. Additional plots are shown in supplemental Figure 2C. (F) Before exposure to attractant supplied by a micropipette containing 10μM fMLP, cells were not pretreated (control), were pretreated with blebbistatin (Blebbis, 100μM, 30 minutes), were infected with lentivirus-containing myosin IIA–targeting shRNAs (MyoII KD), or were pretreated with Y-27632 (30μM, 30 minutes). The 3 images in each row show the positions of individual cells (each identified with a superimposed letter) after the indicated times of exposure to fMLP. White and black arrows point to the poorly developed leading edges and long stretched tails, respectively. Cells infected with virus containing a scramble shRNA exhibited similar response to uninfected control cells. Lower doses of blebbistatin (≤ 50μM) were tested, which required a prolonged period of incubation to exert the same effects as 100μM blebbistatin (data not shown). Scale bar represents 10 μm. Videos of cells with or without treatments are available in supplemental data. (G) DIC kymographs of a dHL-60 cell untreated or treated with inhibitors or myosin IIA shRNAs migrating toward an fMLP (10μM)–containing micropipette. The left panel shows a portion of the neutrophils' leading edge under various conditions. The dotted rectangles indicate the regions of the cell used to generate the kymographs (before fMLP stimulation). The actual lengths of the rectangles are 20 μm in the direction of the arrow. White scale bar represents 1 μm. The right panel shows the DIC kymographs. White scale bar represents 5 μm. In both panels, white arrows indicate the direction of protrusion. Cells a, d, f, and h in panel F were used for the analysis. Approximately 8 minutes of migration was recorded.
Myosin II controls tractions and is necessary for neutrophil chemotaxis. (A) dHL-60 cells were stimulated for 3 minutes by a uniform concentration of 1μM fMLP. Cells were fixed with 3.7% paraformaldehyde and stained with a specific antimyosin heavy chain IIA (MHCIIA) antibody, anti-p[Ser19]MRLC antibody, and rhodamine-conjugated phalloidin to localize filamentous actin (F-actin). The corresponding DIC image is also shown. Scale bar represents 10 μm. (B) dHL-60 cells were transfected with mCherry–myosin IIA (mChe-myoIIA) and stimulated by a micropipette containing 10μM fMLP for the indicated times. mCherry–myosin IIA fluorescence and the corresponding DIC images are shown. Arrows point to the locations of myosin IIA at the leading and trailing edges. Scale bar represents 10 μm. The video of the cell in panel B is available in supplemental data. (C) dHL-60 cells pretreated with blebbistatin (100μM, 30 minutes) were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated elastic polyacrylamide gel for the indicated times. Cells treated with blebbistatin migrated at 1.0 μm/minute on the elastic gel. Traction maps of the cell are shown. Pseudocolor bar representing traction force is given in Pascal (Pa). Scale bar represents 10 μm. The leading edge (within the first 2.2 μm of the cell) is marked by a white line (“Polyacrylamide gel substrates, TFM, and data analysis”). The image series shows part (5.6 seconds) of the whole migratory response. (D) Time series of traction maps from panel C (with 4 additional time points) was analyzed by a customized MATLAB program to determine the average tractions in both leading edge (front) and trailing edge (back) of the cells in a time-dependent manner. The graph shows part (∼ 9.6 seconds) of the whole migratory response. The x-axis indicates time in seconds; y-axis is in Pascal (Pa). (E) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory cell pretreated with blebbistatin (100μM, 30 minutes). The whole migratory response was analyzed. The y-axis represents the power spectral density normalized to the highest peak value (1); x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Cells depleted of myosin IIA exhibited similar response (not shown). Ten cells were analyzed, and a representative cell is shown. Additional plots are shown in supplemental Figure 2C. (F) Before exposure to attractant supplied by a micropipette containing 10μM fMLP, cells were not pretreated (control), were pretreated with blebbistatin (Blebbis, 100μM, 30 minutes), were infected with lentivirus-containing myosin IIA–targeting shRNAs (MyoII KD), or were pretreated with Y-27632 (30μM, 30 minutes). The 3 images in each row show the positions of individual cells (each identified with a superimposed letter) after the indicated times of exposure to fMLP. White and black arrows point to the poorly developed leading edges and long stretched tails, respectively. Cells infected with virus containing a scramble shRNA exhibited similar response to uninfected control cells. Lower doses of blebbistatin (≤ 50μM) were tested, which required a prolonged period of incubation to exert the same effects as 100μM blebbistatin (data not shown). Scale bar represents 10 μm. Videos of cells with or without treatments are available in supplemental data. (G) DIC kymographs of a dHL-60 cell untreated or treated with inhibitors or myosin IIA shRNAs migrating toward an fMLP (10μM)–containing micropipette. The left panel shows a portion of the neutrophils' leading edge under various conditions. The dotted rectangles indicate the regions of the cell used to generate the kymographs (before fMLP stimulation). The actual lengths of the rectangles are 20 μm in the direction of the arrow. White scale bar represents 1 μm. The right panel shows the DIC kymographs. White scale bar represents 5 μm. In both panels, white arrows indicate the direction of protrusion. Cells a, d, f, and h in panel F were used for the analysis. Approximately 8 minutes of migration was recorded.
We next impaired the function of myosin II in neutrophils and assessed the effect on tractions. As expected, inhibiting myosin II with blebbistatin (a specific myosin II inhibitor) in dHL-60 cells and primary neutrophils substantially reduced the tractions at both the leading and the trailing edges by up to 50% (Figure 2C-D; Table 1). Remarkably, myosin II inhibition abolished the periodicity of tractions at both edges: myosin II-inhibited cells no longer exhibited periodic oscillations of tractions but instead showed random fluctuations, as shown by the lack of distinct frequencies/periods in all cells analyzed (n = 10 for dHL-60 cells; n = 6 for primary neutrophils; Figure 2E; supplemental Figure 2C-D). Thus, myosin II is necessary for development and periodicity of tractions in neutrophils and serves as the molecular basis for tractions in neutrophils during chemotaxis.
Inhibition or depletion of myosin II also impaired neutrophil directional migration. In the presence of an fMLP gradient delivered by the micropipette, both dHL-60 cells and primary neutrophils, when plated on FN-coated cover glass, polarized and rapidly migrated in the direction of the pipette with well-developed pseudopodia (Figure 2F; supplemental Figure 5A; supplemental Video 6). In contrast, cells treated with blebbistatin failed to persist in forward movement and formed poorly developed unstable leading edges (Figure 2F; supplemental Video 7; data not shown). DIC kymographs of cells responding to the pipette stimulation demonstrated that blebbistatin-treated cells often retracted their leading edges after initial protrusion, whereas untreated cells exhibited a persistent and progressive pattern of leading-edge protrusion (Figure 2G; supplemental Figure 3A-H). In addition, blebbistatin treatment caused defects in contractility at the back of the cells, which in many cases exhibited long, stretched tails (Figure 2F; supplemental Figure 3D,H), as previously reported.4 Differentiated HL-60 cells in which myosin IIA was depleted by small hairpin RNAs (shRNAs) exhibited similar (albeit less profound) defects (Figure 2F-G; supplemental Figure 3A-D,I; supplemental Video 8). Thus, myosin II inhibition (or depletion) caused neutrophils to form unstable leading edges and long tails. In contrast, inhibition of p160-ROCK, a key component of the previously documented “backness” pathway that regulates neutrophil polarity and chemotaxis,3,4 reduced the level of activated myosin II at the back (Figure 3D), induced neutrophils to form long stretched tails (Figure 2F)26 but failed to affect protrusion of the leading edge (Figure 2F-G; supplemental Figure 3A-H; supplemental Video 9). The backness pathway promotes localized activation of Rho and its downstream kinase p160-ROCK, leading to spatial activation of myosin II at the trailing edge.3,4 Based on the differential effects of myosin II and p160-ROCK inhibition on protrusion, we inferred that myosin II activity at the neutrophil's leading edge is necessary for stabilizing the protrusion response.
![Figure 3. MLCK inhibition/depletion impairs myosin II activity at the leading edge. (A) dHL-60 cells plated on FN-coated coverslips were stimulated for 2 minutes with 1μM fMLP, fixed with 3.7% paraformaldehyde, and stained with a specific anti-MLCK antibody (red) and AlexaFluor-488–conjugated phalloidin (green). The polarized distribution of endogenous MLCK was observed in 188 of 269 polarized cells. (B) A dHL-60 cell treated with ML-7 (25μM, 30 minutes) was exposed to a point source of 10μM fMLP for the times indicated (bottom panel). The cell fails to migrate to the micropipette and shows poorly developed pseudopod (white arrow). An untreated dHL-60 cell (top panel) with well-developed, stable pseudopod is shown. Scale bars represent 10 μm. (C) DIC kymographs (left) of a dHL-60, untreated or treated with ML-7, migrating toward an fMLP (10μM)–containing micropipette. The dotted rectangles indicate the regions of the cell used to generate the kymographs (before fMLP stimulation). The actual lengths of the rectangles are 20 μm in the direction of the arrow. Left panel: Scale bar represents 1 μm. Right panel: Scale bars represent 5 μm. Arrows indicate the direction of protrusion. Five minutes of migration was recorded. The speed of leading-edge protrusion was calculated based on the kymographs (right). The values are mean plus or minus SEM (n = 34 for control, and 32 for cells treated with ML-7). *Value for cells with ML-7 treatment differs from the corresponding control (P < .0001). (D) dHL-60 cells not pretreated or pretreated with ML-7 (25μM, 30 minutes) or Y-27632 (30μM, 30 minutes) were stimulated for 3 minutes by a uniform concentration of 1μM fMLP. Cells were fixed with 3.7% paraformaldehyde and stained with the anti-MHCIIA antibody, anti-p[Ser19]MRLC antibody, and rhodamine-conjugated phalloidin. The corresponding DIC images are also shown. Scale bar represents 10 μm. (E) The distribution of p[Ser19]MRLC in dHL-60 cells with or without ML-7 treatment was analyzed. The mean fluorescence of p[Ser19]MRLC staining at the leading edge and the trailing edge of cells was determined using ImageJ software 1.43, and the ratios between the leading edge and the trailing edge (ie, mean fluorescence intensity at the leading edge/mean fluorescence intensity at the trailing edge) are shown. Values were normalized to the ratio (100%) in control cells and are mean plus or minus SEM (n = 40 for control, and 30 for cells treated with ML-7). Student t tests compared data between experimental groups. *Results significantly different from control (P < .001). (F) Western blot of p[Ser19]MRLC. dHL-60 cells were pretreated with no inhibitors, ML-7 (25μM, 30 minutes), or Y-276322 (30μM, 30 minutes) before exposure to fMLP for 2 minutes in suspension. (Top panel) A typical blot. (Bottom panel) Quantification of blots from 4 separate experiments. Each bar represents the mean plus or minus SEM (error bars). All values were normalized to the signal (100%) detected without the inhibitors. Value for cells treated with M-7 or Y-27632 differs statistically from the control: *P < .01, **P < .001. Total MHCIIA levels were unaltered with the treatments and were used for equal loading in the different lanes. (G) The distribution of total MHCIIA in control dHL-60 cells and cells pretreated with ML-7 or Y-27632 was analyzed. The mean fluorescence of MHCIIA staining at leading and trailing edges was assessed by ImageJ software 1.43, and the ratios between leading and trailing edges are shown. Values were normalized to the ratio (100%) in control cells and are mean plus or minus SEM (n = 40 for control, 30 for cells treated with ML-7, and 25 for cells treated with Y-27632). Results significantly different from control: *P < .05, **P < .001.](/view-large/figure/7113593/zh89991058490003.jpeg)
MLCK inhibition/depletion impairs myosin II activity at the leading edge. (A) dHL-60 cells plated on FN-coated coverslips were stimulated for 2 minutes with 1μM fMLP, fixed with 3.7% paraformaldehyde, and stained with a specific anti-MLCK antibody (red) and AlexaFluor-488–conjugated phalloidin (green). The polarized distribution of endogenous MLCK was observed in 188 of 269 polarized cells. (B) A dHL-60 cell treated with ML-7 (25μM, 30 minutes) was exposed to a point source of 10μM fMLP for the times indicated (bottom panel). The cell fails to migrate to the micropipette and shows poorly developed pseudopod (white arrow). An untreated dHL-60 cell (top panel) with well-developed, stable pseudopod is shown. Scale bars represent 10 μm. (C) DIC kymographs (left) of a dHL-60, untreated or treated with ML-7, migrating toward an fMLP (10μM)–containing micropipette. The dotted rectangles indicate the regions of the cell used to generate the kymographs (before fMLP stimulation). The actual lengths of the rectangles are 20 μm in the direction of the arrow. Left panel: Scale bar represents 1 μm. Right panel: Scale bars represent 5 μm. Arrows indicate the direction of protrusion. Five minutes of migration was recorded. The speed of leading-edge protrusion was calculated based on the kymographs (right). The values are mean plus or minus SEM (n = 34 for control, and 32 for cells treated with ML-7). *Value for cells with ML-7 treatment differs from the corresponding control (P < .0001). (D) dHL-60 cells not pretreated or pretreated with ML-7 (25μM, 30 minutes) or Y-27632 (30μM, 30 minutes) were stimulated for 3 minutes by a uniform concentration of 1μM fMLP. Cells were fixed with 3.7% paraformaldehyde and stained with the anti-MHCIIA antibody, anti-p[Ser19]MRLC antibody, and rhodamine-conjugated phalloidin. The corresponding DIC images are also shown. Scale bar represents 10 μm. (E) The distribution of p[Ser19]MRLC in dHL-60 cells with or without ML-7 treatment was analyzed. The mean fluorescence of p[Ser19]MRLC staining at the leading edge and the trailing edge of cells was determined using ImageJ software 1.43, and the ratios between the leading edge and the trailing edge (ie, mean fluorescence intensity at the leading edge/mean fluorescence intensity at the trailing edge) are shown. Values were normalized to the ratio (100%) in control cells and are mean plus or minus SEM (n = 40 for control, and 30 for cells treated with ML-7). Student t tests compared data between experimental groups. *Results significantly different from control (P < .001). (F) Western blot of p[Ser19]MRLC. dHL-60 cells were pretreated with no inhibitors, ML-7 (25μM, 30 minutes), or Y-276322 (30μM, 30 minutes) before exposure to fMLP for 2 minutes in suspension. (Top panel) A typical blot. (Bottom panel) Quantification of blots from 4 separate experiments. Each bar represents the mean plus or minus SEM (error bars). All values were normalized to the signal (100%) detected without the inhibitors. Value for cells treated with M-7 or Y-27632 differs statistically from the control: *P < .01, **P < .001. Total MHCIIA levels were unaltered with the treatments and were used for equal loading in the different lanes. (G) The distribution of total MHCIIA in control dHL-60 cells and cells pretreated with ML-7 or Y-27632 was analyzed. The mean fluorescence of MHCIIA staining at leading and trailing edges was assessed by ImageJ software 1.43, and the ratios between leading and trailing edges are shown. Values were normalized to the ratio (100%) in control cells and are mean plus or minus SEM (n = 40 for control, 30 for cells treated with ML-7, and 25 for cells treated with Y-27632). Results significantly different from control: *P < .05, **P < .001.
MLCK inhibition/depletion impairs myosin II activity at the leading edge. (A) dHL-60 cells plated on FN-coated coverslips were stimulated for 2 minutes with 1μM fMLP, fixed with 3.7% paraformaldehyde, and stained with a specific anti-MLCK antibody (red) and AlexaFluor-488–conjugated phalloidin (green). The polarized distribution of endogenous MLCK was observed in 188 of 269 polarized cells. (B) A dHL-60 cell treated with ML-7 (25μM, 30 minutes) was exposed to a point source of 10μM fMLP for the times indicated (bottom panel). The cell fails to migrate to the micropipette and shows poorly developed pseudopod (white arrow). An untreated dHL-60 cell (top panel) with well-developed, stable pseudopod is shown. Scale bars represent 10 μm. (C) DIC kymographs (left) of a dHL-60, untreated or treated with ML-7, migrating toward an fMLP (10μM)–containing micropipette. The dotted rectangles indicate the regions of the cell used to generate the kymographs (before fMLP stimulation). The actual lengths of the rectangles are 20 μm in the direction of the arrow. Left panel: Scale bar represents 1 μm. Right panel: Scale bars represent 5 μm. Arrows indicate the direction of protrusion. Five minutes of migration was recorded. The speed of leading-edge protrusion was calculated based on the kymographs (right). The values are mean plus or minus SEM (n = 34 for control, and 32 for cells treated with ML-7). *Value for cells with ML-7 treatment differs from the corresponding control (P < .0001). (D) dHL-60 cells not pretreated or pretreated with ML-7 (25μM, 30 minutes) or Y-27632 (30μM, 30 minutes) were stimulated for 3 minutes by a uniform concentration of 1μM fMLP. Cells were fixed with 3.7% paraformaldehyde and stained with the anti-MHCIIA antibody, anti-p[Ser19]MRLC antibody, and rhodamine-conjugated phalloidin. The corresponding DIC images are also shown. Scale bar represents 10 μm. (E) The distribution of p[Ser19]MRLC in dHL-60 cells with or without ML-7 treatment was analyzed. The mean fluorescence of p[Ser19]MRLC staining at the leading edge and the trailing edge of cells was determined using ImageJ software 1.43, and the ratios between the leading edge and the trailing edge (ie, mean fluorescence intensity at the leading edge/mean fluorescence intensity at the trailing edge) are shown. Values were normalized to the ratio (100%) in control cells and are mean plus or minus SEM (n = 40 for control, and 30 for cells treated with ML-7). Student t tests compared data between experimental groups. *Results significantly different from control (P < .001). (F) Western blot of p[Ser19]MRLC. dHL-60 cells were pretreated with no inhibitors, ML-7 (25μM, 30 minutes), or Y-276322 (30μM, 30 minutes) before exposure to fMLP for 2 minutes in suspension. (Top panel) A typical blot. (Bottom panel) Quantification of blots from 4 separate experiments. Each bar represents the mean plus or minus SEM (error bars). All values were normalized to the signal (100%) detected without the inhibitors. Value for cells treated with M-7 or Y-27632 differs statistically from the control: *P < .01, **P < .001. Total MHCIIA levels were unaltered with the treatments and were used for equal loading in the different lanes. (G) The distribution of total MHCIIA in control dHL-60 cells and cells pretreated with ML-7 or Y-27632 was analyzed. The mean fluorescence of MHCIIA staining at leading and trailing edges was assessed by ImageJ software 1.43, and the ratios between leading and trailing edges are shown. Values were normalized to the ratio (100%) in control cells and are mean plus or minus SEM (n = 40 for control, 30 for cells treated with ML-7, and 25 for cells treated with Y-27632). Results significantly different from control: *P < .05, **P < .001.
MLCK regulates leading edge stability, myosin II activity, and tractions
We next explored how localization-specific myosin II activity and tractions were controlled at the leading and the trailing edge. Earlier studies demonstrated that the spatial activation of myosin II at the trailing edge is dependent on p160-ROCK and necessary for tail retraction and de-adhesion.3,4 We thus sought to understand how myosin II was spatially activated at the leading edge in neutrophils. MLCK, a [Ca2+]/calmodulin-dependent kinase, phosphorylates MRLC and induces contractility in multiple processes,27 including cell motility.28,29 To ask whether MLCK spatially regulated myosin II activation at the leading edge of neutrophils, we first examined its expression and subcellular localization. Western blotting indicated that both primary human neutrophils and dHL-60 cells mainly expressed the approximately 130-kDa short isoform of MLCK (supplemental data; supplemental Figure 4A). In nonstimulated neutrophils, MLCK was diffusely distributed in the cytosol, with some cortical localization (Figure 3A). When stimulated with fMLP, it was recruited to the leading edge of polarized cells and colocalized with F-actin (Figure 3A). The polarized recruitment was confirmed by ectopic expression of EGFP-tagged short isoform MLCK (sMLCK-EGFP, in accordance with its expression in neutrophils; supplemental data; supplemental Figure 4B-C; supplemental Videos 10-11). MLCK inhibition by a specific inhibitor ML-7 (25μM) impaired the stability of the leading edge and markedly reduced the speed of protrusion of dHL-60 cells and primary neutrophils, as shown by experiments with the micropipette assay (Figure 3B-C; supplemental Figure 5). Similar to myosin II inhibition, MLCK inhibition caused neutrophils to often retract their leading edges during migration (∼ 0.4/minute and 0.03/minute for treated and untreated dHL-60 cells, respectively; ∼ 0.7/minute and 0.15/minute for treated and untreated primary cells, respectively). However, unlike myosin II inhibition, MLCK inhibition failed to induce formation of stretched tails at the back of the cells (Figure 3B; supplemental Figure 5A).
In circulating neutrophils, β2-integrins are highly expressed and mediate the interactions with the endothelial cells. We thus assessed whether MLCK was also required for chemotaxis of neutrophils stimulated on ECM substrates for β2-integrins. Differentiated HL-60 cells, when stimulated by an fMLP gradient on fibrinogen or intercellular adhesion molecule-1 (supplemental Figure 6; data not shown), rapidly polarized and migrated toward the source of the chemoattractants (supplemental Figure 6). ML-7 treatment markedly impaired the leading edge stability of these cells (supplemental Figure 6). These results suggest that MLCK might play a conserved role in the regulation of neutrophil polarity and chemotaxis under different microenvironmental settings.
The specificity of ML-7 for MLCK, instead of for other protein kinase, including protein kinase A, was demonstrated at a similar concentration.30 Nevertheless, to further confirm the specificity of ML-7, we used RNAi-mediated knockdown to deplete MLCK in dHL-60 cells. Depletion of MLCK led to similar defects (supplemental data; supplemental Figure 7A-D). In addition, consistent with their effects on the leading edges, MLCK inhibition and depletion prevented accumulation of polymerized actin, 3′-phosphoinositol lipids (PI3Ps), and Rac-GTP at the leading edge (supplemental data; supplemental Figures 7E, 8). Actin polymers, PI3Ps, and Rac-GTP are key components of the “frontness” pathway and play essential roles in establishing the leading edge during neutrophil chemotaxis.1,4 Thus, our results show that MLCK is spatially recruited to the leading edge and is necessary for leading edge stability during neutrophil chemotaxis.
MRLCs are the only known substrate for MLCK.27 The asymmetric distribution of MLCK prompted us to ask whether this kinase would selectively control the activity of myosin II at the leading edge. The ratio of mean immunofluorescence intensity of p[19]-MRLC between the leading and the trailing edges was approximately 25% lower in ML-7–treated dHL-60 cells than in the control cells (Figure 3D-E), suggesting that MLCK inhibition impaired the accumulation of activated myosin II at the leading edge compared with the rest of the cell body. The use of ratio of mean fluorescence intensity discounts the variations in the levels of fluorescence probes among cells and is thus more appropriate for assessing the relative accumulation of the fluorescent signals. Furthermore, treatment with the p160-ROCK inhibitor Y-27632 sharply reduced the distribution of phosphorylated myosin II at the trailing edge (Figure 3D), in keeping with an earlier report.4 Inhibition of myosin II activity by ML-7 and Y-27632 was also demonstrated by Western blotting of total p[19]-MRLC levels (Figure 3F). Intriguingly, both ML-7 and Y-27632 treatments increased the relative distribution of myosin IIA protein at the leading edge (Figure 3G; 24% and 41%, respectively), probably the result of the alterations in overall cytoskeletal organization. Taking into account the increased distribution of myosin IIA at the front, we inferred that ML-7 caused more than 30% decrease in the relative level of activated myosin II (ie, p[19]-MRLC/total myosin IIA) at the leading edge.
Inhibiting or depleting MLCK in neutrophils markedly reduced the tractions at the leading edge (Figure 4A-B; supplemental Video 12) in cells migrating on the substrate with stiffness of 3.5 kPa (Table 1). Interestingly, the same treatments also reduced the tractions at the trailing edge (Figure 4A-B; Table 1). Furthermore, MLCK inhibition (or depletion) abolished the periodic pattern of tractions at both the leading and the trailing edge in dHL-60 cells and primary neutrophils (Figure 4B-C; supplemental Figures 9A-B, 10A). Because the tractions in MLCK-inhibited/depleted cells were reduced at both edges and were no longer periodic, this finding suggested coordination and coupling of tractions at the front and the back under this matrix-stiffness condition. In keeping with this notion, treatment of cells with Y-27632 also substantially reduced the tractions and prevented the periodic oscillations at both the leading and the trailing edges (Figure 4D-F; supplemental Figures 9C, 10B; Table 1).

Localization-specific myosin activities are necessary for tractions in neutrophils. (A) dHL-60 cells depleted of MLCK were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated elastic polyacrylamide gel for the indicated times. Traction maps of the cell are shown. Pseudocolor bar representing tractions is given in Pascal (Pa). Scale bar represents 5 μm. The leading edge (within the first 2.2 μm of the cell) is marked by a white line. The image series shows part (5.6 seconds) of the whole migratory response. Cells treated with ML-7 exhibited similar responses. Cells treated with ML-7 or depleted of MLCK migrated at 1.1 μm/minute on the elastic gel. The video of the cell in panel A is available in supplemental data. (B) Time series of traction maps from panel A (with 5 additional time points) was analyzed by a customized MATLAB program to determine the average traction force in both leading edge (front) and trailing edge (back) of the cells in a time-dependent manner. The graph shows part (∼ 9.6 seconds) of the whole migratory response. The x-axis indicates time in seconds; y-axis is in Pascal (Pa). (C) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory cell depleted of MLCK. The whole migratory response was analyzed. The y-axis represents the power spectral density normalized to the highest peak value (1); x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Cells pretreated with ML-7 exhibited similar response (not shown). Ten cells were analyzed, and a representative cell is shown. Additional plots for ML-7 treatment and MLCK depletion are shown in supplemental Figure 9A and B. (D) dHL-60 cells pretreated with Y-27632 (30 μm, 30 minutes) were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated elastic polyacrylamide gel for the indicated times. Traction force maps of the cell are shown. Pseudocolor bar representing traction force is given in Pascal (Pa). Scale bar represents 10 μm. The leading edge (within the first 3 μm of the cell) is marked by a white line (“Polyacrylamide gel substrates, TFM, and data analysis”). The image series shows part (5.6 seconds) of the whole migratory response. (E) Time series of traction maps from panel D (with 5 additional time points) was analyzed by a customized MATLAB program to determine the average traction force in both leading edge (front) and trailing edge (back) of the cells in a time-dependent manner. The graph shows part (∼ 9.6 seconds) of the whole migratory response. The x-axis indicates time in seconds; y-axis is in Pascal (Pa). (F) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory cell pretreated with Y-27632. The whole migratory response was analyzed. The y-axis represents the power spectral density normalized to the highest peak value (1); x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Eight cells were analyzed, and a representative cell is shown. Additional plots are shown in supplemental Figure 9C.
Localization-specific myosin activities are necessary for tractions in neutrophils. (A) dHL-60 cells depleted of MLCK were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated elastic polyacrylamide gel for the indicated times. Traction maps of the cell are shown. Pseudocolor bar representing tractions is given in Pascal (Pa). Scale bar represents 5 μm. The leading edge (within the first 2.2 μm of the cell) is marked by a white line. The image series shows part (5.6 seconds) of the whole migratory response. Cells treated with ML-7 exhibited similar responses. Cells treated with ML-7 or depleted of MLCK migrated at 1.1 μm/minute on the elastic gel. The video of the cell in panel A is available in supplemental data. (B) Time series of traction maps from panel A (with 5 additional time points) was analyzed by a customized MATLAB program to determine the average traction force in both leading edge (front) and trailing edge (back) of the cells in a time-dependent manner. The graph shows part (∼ 9.6 seconds) of the whole migratory response. The x-axis indicates time in seconds; y-axis is in Pascal (Pa). (C) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory cell depleted of MLCK. The whole migratory response was analyzed. The y-axis represents the power spectral density normalized to the highest peak value (1); x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Cells pretreated with ML-7 exhibited similar response (not shown). Ten cells were analyzed, and a representative cell is shown. Additional plots for ML-7 treatment and MLCK depletion are shown in supplemental Figure 9A and B. (D) dHL-60 cells pretreated with Y-27632 (30 μm, 30 minutes) were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated elastic polyacrylamide gel for the indicated times. Traction force maps of the cell are shown. Pseudocolor bar representing traction force is given in Pascal (Pa). Scale bar represents 10 μm. The leading edge (within the first 3 μm of the cell) is marked by a white line (“Polyacrylamide gel substrates, TFM, and data analysis”). The image series shows part (5.6 seconds) of the whole migratory response. (E) Time series of traction maps from panel D (with 5 additional time points) was analyzed by a customized MATLAB program to determine the average traction force in both leading edge (front) and trailing edge (back) of the cells in a time-dependent manner. The graph shows part (∼ 9.6 seconds) of the whole migratory response. The x-axis indicates time in seconds; y-axis is in Pascal (Pa). (F) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory cell pretreated with Y-27632. The whole migratory response was analyzed. The y-axis represents the power spectral density normalized to the highest peak value (1); x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Eight cells were analyzed, and a representative cell is shown. Additional plots are shown in supplemental Figure 9C.
It was recently documented that the chemotactic behaviors of neutrophils were influenced by substrate stiffness.12 To investigate whether MLCK played a more conserved role in the regulation of tractions, we assessed the effect of MLCK inhibition in neutrophils migrating on a stiffer substrate (100 kPa). Primary neutrophils migrating toward an fMLP-containing micropipette exerted higher tractions under this condition, in keeping with the previous report12 (Table 1). Tractions at the leading and the trailing edges both demonstrated periodicity (4.7 ± 1.5 seconds and 4.7 ± 1.3 seconds, respectively) and were out of phase by 1.0 plus or minus 0.3 seconds (Figure 5A-B). Myosin II inhibition, as expected, markedly reduced the levels and prevented the periodicity of traction at both edges (supplemental Figure 11A). MLCK inhibition exerted very similar effects on tractions to blebbistatin treatment (supplemental Figure 11B). Intriguingly, although treatment of cells with Y-27632 reduced tractions by 40% to 45% (Table 1), the cells nevertheless exhibited periodic tractions at both edges (4.7 ± 0.9 seconds and 5.0 ± 1.3 seconds, respectively; Figure 5C), with a lag of 0.90 plus or minus 0.33 seconds (Figure 5D). These results suggest a differential regulatory pattern of tractions in cells migrating on the stiffer substrate.

The pattern and regulation of tractions in neutrophils migrating on a stiffer substrate. (A) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory primary neutrophil. PSD plots were generated based on the results from Fourier analysis of the traction values. The y-axis represents the PSD normalized to the highest peak value (1). The x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Primary cells were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated polyacrylamide gel (100 kPa) for 4 to 5 minutes. Six cells were analyzed, and a representative cell is shown. (B) Left panel: Cross-correlation between tractions at the leading edge and the trailing edge against time offset during migration for individual primary neutrophils. Dotted lines indicate zero offset. Data from 3 representative cells are shown. Time bar represents 24 seconds. Right panel: Summary of time offsets between leading edges and trailing edges (n = 6 cells) in primary cells. (C) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory primary neutrophil pretreated with Y-27632 (30μM, 30 minutes). PSD plots were generated based on the results from Fourier analysis of the traction values. The y-axis represents the PSD normalized to the highest peak value (1). The x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Cells were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated polyacrylamide gel (100 kPa) for 4 to 5 minutes. Six cells were analyzed, and a representative cell is shown. (D) Left panel: Cross-correlation between tractions at the leading edge and the trailing edge against time offset during migration for individual Y-27632–treated primary neutrophils. Dotted lines indicate zero offset. Data from 3 representative cells are shown. Time bar represents 24 seconds. Right panel: Summary of time offsets between leading edges and trailing edges (n = 6 cells) in Y-27632–treated primary cells.
The pattern and regulation of tractions in neutrophils migrating on a stiffer substrate. (A) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory primary neutrophil. PSD plots were generated based on the results from Fourier analysis of the traction values. The y-axis represents the PSD normalized to the highest peak value (1). The x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Primary cells were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated polyacrylamide gel (100 kPa) for 4 to 5 minutes. Six cells were analyzed, and a representative cell is shown. (B) Left panel: Cross-correlation between tractions at the leading edge and the trailing edge against time offset during migration for individual primary neutrophils. Dotted lines indicate zero offset. Data from 3 representative cells are shown. Time bar represents 24 seconds. Right panel: Summary of time offsets between leading edges and trailing edges (n = 6 cells) in primary cells. (C) PSD plots of tractions at the leading edge (left panel) and the trailing edge (right panel) of a migratory primary neutrophil pretreated with Y-27632 (30μM, 30 minutes). PSD plots were generated based on the results from Fourier analysis of the traction values. The y-axis represents the PSD normalized to the highest peak value (1). The x-axis shows the oscillation frequency (Hertz; top) or period (seconds; bottom). Cells were allowed to migrate toward chemoattractant-containing micropipette (fMLP, 10μM) on a FN-coated polyacrylamide gel (100 kPa) for 4 to 5 minutes. Six cells were analyzed, and a representative cell is shown. (D) Left panel: Cross-correlation between tractions at the leading edge and the trailing edge against time offset during migration for individual Y-27632–treated primary neutrophils. Dotted lines indicate zero offset. Data from 3 representative cells are shown. Time bar represents 24 seconds. Right panel: Summary of time offsets between leading edges and trailing edges (n = 6 cells) in Y-27632–treated primary cells.
MLCK regulates leading edge adhesion by activating α5β1-integrin
How does MLCK-mediated myosin contractility control leading-edge stability in neutrophils? During migration, protrusion and adhesion of the leading edge are tightly coupled.31 Cell adhesion sites are required to stabilize leading edges and promote cell polarity in cells migrating on flat surfaces.31 MLCK-mediated myosin II contractility may be necessary for leading edge protrusion. Alternatively, the defects of MLCK inhibition (or depletion) on neutrophil polarity can be interpreted as the inability of the cells to attach their protrusive pseudopods, resulting in unstable and poorly developed leading edges. Indeed, blocking leading edge adhesion of neutrophils with Arg-Gly-Asp (RGD) peptides targeting α5β1-integrin, the main FN receptors in neutrophils, caused defects highly reminiscent of those induced by MLCK inhibition (supplemental data; supplemental Figure 12).
MLCK inhibition markedly impaired neutrophil adhesion. First, experiments with a commonly used cell plating assay demonstrated that MLCK inhibition compromised adhesion of dHL-60 cells to FN (Figure 6A). Interestingly, MLCK inhibition specifically prevented cell adhesion induced by fMLP because ML-7 had little effect on cell adhesion to FN in the absence of fMLP stimulation (Figure 6A). To further analyze adhesion at the single-cell level, we used TIRF microscopy in dHL-60 cells expressing GFP-tagged α5-integrin. The goal of this experiment was to assess whether MLCK inhibition impaired α5β1-integrin-mediated leading edge attachment. The evanescent wave causes excitation of fluorescent molecules in an optical section (< 100 nm) without exciting molecules throughout the specimen and allows visualization of signaling activity in living cells in contact with the coverglass (substrate). In response to a point source of fMLP gradient, neutrophils extended a protrusive leading edge and established contact with the FN substrate, as indicated by the emergence of GFP fluorescence at the leading edge (supplemental Figure 13A). In addition, de-adhesion and retraction of trailing edge were apparent, as suggested by the disappearance of GFP fluorescence at the trailing edge of migrating cells (supplemental Figure 13A). MLCK inhibition markedly impaired leading edge attachment, as suggested by the absence or reduction of fluorescent signals of GFP–α5-integrin at the leading edge (supplemental Figure 13B). We analyzed attachment quantitatively in control and MLCK-inhibited cells by measuring TIRF signals within the leading edge (as defined by DIC images; Figure 6B). This analysis suggested that MLCK inhibition reduced leading edge adhesion by 78% (Figure 6B). In contrast, the same treatment only slightly altered attachment of the area outside the leading edge (denoted as Cell body; Figure 6B right panel). Thus, MLCK inhibition impaired leading edge adhesion. However, these results alone could not distinguish whether MLCK mediates leading edge protrusion or adhesion, as discussed in the first paragraph of this subsection. As expected, cells treated with Y-27632 exhibited long stretched tail that failed to retract properly, whereas cells with blebbistatin treatment showed defects in both leading edge attachment and tail retraction (supplemental Figure 13C-D).

MLCK controls neutrophil adhesion and integrin activation. (A) dHL-60 cells pretreated with (25μM, 30 minutes) or without ML-7 were not stimulated or stimulated in suspension by 1μM fMLP and allowed to adhere to FN-coated surface for 30 minutes, after which the degree of cell adhesion was assessed. Values were normalized to adhesion in control cells (100%) with fMLP and are mean plus or minus SEM (n = 4). *Results significantly different from control (P < .001). (B) Assessment of adhesion area in leading edge and cell body in control and ML-7–treated cells transfected with EGFP-α5-integrin and exposed to a point source of 10μM fMLP. Cell images were from supplemental Figure 13. Left panel: The time point at which leading edge protrusion of individual cells was maximal was selected for analysis. Leading edge (denoted as “L”) in TIRF image was demarcated by the corresponding DIC image, and the rest of the cell was defined as cell body (denoted as “C”). Right panel: Fluorescence intensities of the leading edge and the cell body in the TIRF images of control and ML-7–treated cells were determined with ImageJ software 1.43, and the resulting values were used to quantify cell attachment of the both leading edge and the cell body. The plot shows the relative values in each region compared with control (100%) in the presence of fMLP stimulation. Values are mean plus or minus SEM (n = 13 for control, 11 for cells treated with ML-7). *Results significantly different from control (P < .0001). (C-D) Top panel: Localization of activated α5β1-integrins in polarized neutrophils pretreated with or without ML-7 (25μM, 30 minutes). dHL-60 cells pretreated with or without ML-7 were plated on FN-coated coverslips and stimulated for 3 minutes by 1μM fMLP. After washing, cells were fixed with 3.7% paraformaldehyde, incubated with GST-FN III9-11 (50 μg/mL) for 15 minutes at 37°C, and stained with an anti-GST antibody and anti–α5-integrin antibody. Fluorescent and phase-contrast images were collected using confocal fluorescence microscopy. The images of a representative cell for each condition are shown (n = 20 for control and n = 17 for ML-7 treatment). Arrows indicate the leading edge. White line indicates the path along which line profile was obtained. The weak signals for GST-FN III9-11 binding may be attributed to the relative low levels of activated α5β1-integrin in the cells. Scale bar represents 10 μm. Bottom panel: Line profiles of GST-FN III9-11 and α5-integrin fluorescence in cells shown in the top panel. The graphs plot fluorescence intensity of each protein (y-axis; in arbitrary units) versus distance (x-axis in pixels) along the white line on the phase-contrast image of the cells. (E-F) Levels of activated α5β1-integrins in cells with or without ML-7 treatment. dHL-60 cells pretreated with or without ML-7 were stimulated by a uniform concentration of fMLP (1μM) in either suspension (E) or adhesion conditions (F). A typical blot is shown on the left. (Right panel) Quantification of blots from 4 separate experiments. The y-axis represents relative intensities (measured with ImageJ software 1.43) with values normalized to the signal (1) detected in the control cells without ML-7 treatment. Each bar represents the mean plus or minus SEM (n = 4). Results significantly different from those of cells without fMLP stimulation: *P < .0001, **P < .001.
MLCK controls neutrophil adhesion and integrin activation. (A) dHL-60 cells pretreated with (25μM, 30 minutes) or without ML-7 were not stimulated or stimulated in suspension by 1μM fMLP and allowed to adhere to FN-coated surface for 30 minutes, after which the degree of cell adhesion was assessed. Values were normalized to adhesion in control cells (100%) with fMLP and are mean plus or minus SEM (n = 4). *Results significantly different from control (P < .001). (B) Assessment of adhesion area in leading edge and cell body in control and ML-7–treated cells transfected with EGFP-α5-integrin and exposed to a point source of 10μM fMLP. Cell images were from supplemental Figure 13. Left panel: The time point at which leading edge protrusion of individual cells was maximal was selected for analysis. Leading edge (denoted as “L”) in TIRF image was demarcated by the corresponding DIC image, and the rest of the cell was defined as cell body (denoted as “C”). Right panel: Fluorescence intensities of the leading edge and the cell body in the TIRF images of control and ML-7–treated cells were determined with ImageJ software 1.43, and the resulting values were used to quantify cell attachment of the both leading edge and the cell body. The plot shows the relative values in each region compared with control (100%) in the presence of fMLP stimulation. Values are mean plus or minus SEM (n = 13 for control, 11 for cells treated with ML-7). *Results significantly different from control (P < .0001). (C-D) Top panel: Localization of activated α5β1-integrins in polarized neutrophils pretreated with or without ML-7 (25μM, 30 minutes). dHL-60 cells pretreated with or without ML-7 were plated on FN-coated coverslips and stimulated for 3 minutes by 1μM fMLP. After washing, cells were fixed with 3.7% paraformaldehyde, incubated with GST-FN III9-11 (50 μg/mL) for 15 minutes at 37°C, and stained with an anti-GST antibody and anti–α5-integrin antibody. Fluorescent and phase-contrast images were collected using confocal fluorescence microscopy. The images of a representative cell for each condition are shown (n = 20 for control and n = 17 for ML-7 treatment). Arrows indicate the leading edge. White line indicates the path along which line profile was obtained. The weak signals for GST-FN III9-11 binding may be attributed to the relative low levels of activated α5β1-integrin in the cells. Scale bar represents 10 μm. Bottom panel: Line profiles of GST-FN III9-11 and α5-integrin fluorescence in cells shown in the top panel. The graphs plot fluorescence intensity of each protein (y-axis; in arbitrary units) versus distance (x-axis in pixels) along the white line on the phase-contrast image of the cells. (E-F) Levels of activated α5β1-integrins in cells with or without ML-7 treatment. dHL-60 cells pretreated with or without ML-7 were stimulated by a uniform concentration of fMLP (1μM) in either suspension (E) or adhesion conditions (F). A typical blot is shown on the left. (Right panel) Quantification of blots from 4 separate experiments. The y-axis represents relative intensities (measured with ImageJ software 1.43) with values normalized to the signal (1) detected in the control cells without ML-7 treatment. Each bar represents the mean plus or minus SEM (n = 4). Results significantly different from those of cells without fMLP stimulation: *P < .0001, **P < .001.
We next asked whether MLCK was necessary for leading edge protrusion. We took advantage of a feature of neutrophils: their ability to polarize when stimulated with chemoattractants, even in suspension. When exposed to fMLP, neutrophils in suspension quickly (within 1-2 minutes) establish a morphologic leading edge.32 The leading edge does not persist and retracts after stimulation.2,33-35 Attractant stimulation of neutrophils in suspension also suffices to lead to accumulation of polymerized actin, PI3Ps, and Rac-GTP, albeit transiently, consistent with the morphologic response.1,2,4 When held with a pipette, dHL-60 cells responded to a gradient of fMLP from an adjacent pipette by extending a leading edge (supplemental Figure 14A). The leading edge continued to grow and then retracted. Cells treated with ML-7 exhibited no detectable defects in the protrusive behavior and dynamics when stimulated with the fMLP gradient (supplemental Figure 14A-B). We next measured the level of polymerized actin, Rac-GTP, and phospho-Akt (activated Akt, readout for PI3Ps)2,4 with or without fMLP stimulation. Consistent with previous findings,1,2,4 fMLP addition induced a rapid and transient accumulation of polymerized actin, phospho-Akt, and Rac-GTP in neutrophils in suspension, which peaked at 30 to 60 seconds after stimulation (supplemental Figure 14C-E). Treatment with ML-7 failed to prevent any of these frontness markers in cells exposed to fMLP in suspension (supplemental Figure 14C-E). Therefore, MLCK is not required for protrusion per se but appears to regulate leading edge adhesion. In addition, because MLCK inhibition/depletion impaired the leading edge accumulation of PI3Ps, Rac-GTP, and actin polymers in adherent cells (supplemental Figures 7E, 8), these results imply that cell adhesion is necessary for the stability of the frontness signals.
Integrin activation refers to a switch from a low-affinity to a high-affinity state of ligand binding. Integrin activation is the key step for cell adhesion to the ECM.36 To explain how MLCK controls cell adhesion during neutrophil chemotaxis, we tested whether MLCK is involved in integrin activation during chemotaxis. We assessed the localization and expression of activated α5β1-integrin with a GST-tagged protein containing the GST-FN III9-11, which specifically binds active α5β1-integrin.14 Although total α5β1-integrin (as detected by anti–α5-integrin antibody) was found evenly distributed throughout the cell, activated α5β1-integrin was enriched at the leading edge of polarized dHL-60 cells (Figure 6C; a focal plane from the confocal microscopy analysis is shown). Consistently, the line profile of the polarized cell exhibited the highest level of GST-FN III9-11 binding at the leading edge (Figure 6C). MLCK inhibition caused the level of active α5β1-integrin to markedly reduce at the leading edge, whereas the localization pattern of total α5β1-integrin remained unchanged (Figure 6D). Incubation of cells with GST alone failed to produce fluorescent signals (supplemental Figure 14F). These results suggest that MLCK controls α5β1-integrin activity at the neutrophil's leading edge.
We next assessed the level of activated α5β1-integrin in cells with or without MLCK inhibition biochemically. As shown in Figure 6E, ML-7 exerted little effect on fMLP-induced activation in neutrophils in suspension, indicating that MLCK activity is not necessary for the inside-out activation of α5β1-integrin by fMLP (ie, in the absence of adhesion to ECM). However, the same treatment prevented α5β1-integrin activation when cells were attached to the FN substrate (Figure 6F), demonstrating that MLCK is necessary for α5β1-integrin activation in adherent cells. The dependence of MLCK-mediated integrin activity on cell-ECM adhesion suggested that tractions may play a special role in this regulation. Traction-induced cytoskeletal tension could activate integrin mechanically, as discussed in the last paragraph in “Discussion.”
Discussion
Taken together, our results reveal novel mechanical aspects of cell polarity and motility in neutrophils and are summarized in a model (Figure 7). We discovered a periodic pattern of tractions at the leading and the trailing edges in a neutrophil-like cell line and primary neutrophils during chemotaxis. This oscillatory pattern has not been documented for any biochemical or mechanical factors in neutrophils during chemotaxis. What might be the function of the periodic tractions? We speculate that the oscillations at the front of the cell may be linked to the cycle of leading edge extension and adhesion. In this scenario, extension of the pseudopod occurs with minimal contact with the ECM substrate and is thus associated with weak tractions. In contrast, strong tractions could result from establishment and stabilization of leading edge attachment. In addition, the traction oscillations at the cell's rear may correspond to periodic contractions that enable the cell to pull the trailing edge forward. Interestingly, in addition to the periodic patterns, we found that the traction oscillations at the front and the back are out of phase, with the tractions at the front leading those at the back, thus pointing to a timing mechanism that allows neutrophils to coordinate leading edge adhesion and trailing edge de-adhesion to ensure persistent movements. We can only speculate about the molecular mechanism that gives rise to the temporal shift. Chemoattractant-induced activation of MLCK may precede that of p160-ROCK (Figure 7), which could cause the delay in myosin II activation and myosin II contractility at the rear. This possibility could be tested by examining the temporal dynamics of MLCK, p160-ROCK, and Rho activation in response to attractants, probably with high-sensitivity live cell biosensors for MLCK and p160-ROCK activities. Notably, our findings contrasted an earlier study in which traction stresses were detected mostly in the uropod of neutrophils migrating on a stiffer substrate (9 kPa).37 The differential patterns of tractions observed may be attributed to the differences in substrate stiffness, temporal resolutions of the studies, and/or other undefined factors.

Integration of mechanical and biochemical signals to regulate leading edge adhesion and trailing edge de-adhesion: a model. The model is proposed based on analyses of cell migration on the 3.5-kPa substrate. GPCR indicates G protein-coupled receptor.
Integration of mechanical and biochemical signals to regulate leading edge adhesion and trailing edge de-adhesion: a model. The model is proposed based on analyses of cell migration on the 3.5-kPa substrate. GPCR indicates G protein-coupled receptor.
How are tractions regulated in neutrophils during chemotaxis? Our current findings suggest that tractions at the leading and the trailing edges in neutrophils require myosin II activation. Inhibition of myosin II impairs the level and the periodicity of tractions. Our results suggest that the recruitment of MLCK to the neutrophil's leading edge activates MRLC and myosin II contractility, allowing cells to exert tractions on the ECM substrate, enhance integrin activity, and stabilize leading edge adhesion (Figure 7). The spatially controlled activation of Rho, p160-ROCK, and subsequent myosin II activation4 leads to contraction of actin-myosin complexes at the trailing edge, causing it to de-adhere (Figure 7). At least 3 possibilities, alone or in combination, could account for the periodic pattern of tractions. It might be that myosin II is activated periodically during chemotaxis. Alternatively, the strength of cell-substrate adhesion might be temporally regulated, leading to alternation of weak and strong local tractions during neutrophil migration. Third, there might be myosin II-independent mechanism(s) that might also contribute to the development and periodicity of tractions. In our experiments, there are significant amounts of tractions remaining after inhibition of total or localization-specific myosin II activities. Our results are in keeping with a recent report demonstrating that inhibition of myosin II functions fails to abolish tractions in Dictyostelium cells during chemotaxis.38 One group of putative candidates that might control tractions in neutrophils are plasma membrane-anchored myosin class I molecules, which reportedly could generate membrane tension by pulling on F-actin coupled to ECM via integrins.39 Other candidates include force-bearing cytoskeletal molecules, such as microtubules and intermediate filaments that can transmit force to actin through various intercytoskeletal linkage.40,41 More complete understanding of the detailed mechanisms governing the periodic and out-of-phase behavior of tractions awaits future experiments. In addition, future experiments should investigate whether the observed spatiotemporal dynamics of tractions and its regulatory pattern can be extended to neutrophils under other extracellular settings (eg, on ECM substrates for β2-integrin) and to other amoeboid cells.
In slow-migrating cells, such as fibroblasts, MLCK is mainly found in stress fibers.42 Distribution of MLCK in neutrophils differs from this pattern. As in neutrophils, MLCK and ROCK in fibroblasts play distinct roles in regulating membrane protrusions and adhesion dynamics during cell migration.43 MLCK inhibition blocks MRLC phosphorylation at the cell periphery, but not in the center, and prevents zyxin-containing adhesions at the periphery. These cells generate membrane protrusions all around the cell, turn more frequently, and migrate less effectively. In contrast, ROCK inhibition blocks MRLC phosphorylation in the center and prevents formation of focal adhesions. These cells move faster and straighter. Thus, although MRLC activation in fibroblasts is spatially controlled by MLCK and ROCK, their distribution and function in the control of cell migration seem to differ from neutrophils.
How might MLCK inhibition (or depletion) at the leading edge affect tractions at both edges in neutrophils during chemotaxis? It is well accepted that organization of actin cytoskeleton in cells is heavily dependent on mechanical force. Increasing tension in the actin network leads to formation of actin bundles, which can be prevented by the inhibition of myosin activity.44,45 More recently, it was demonstrated that nonmuscle myosin II is required, not only to establish but also to maintain integrity of the actomyosin network.46 Based on these results, we speculate that the global effect of MLCK inhibition on tractions in neutrophils might be linked to its ability to regulate myosin II-dependent contractility. In this scenario, depletion or inhibition of MLCK impairs myosin II contractility at the leading edge and in turn disrupts the structure and organization of the actin cytoskeleton (Figure 7), as shown by experiments with live and fixed neutrophils. Myosin II might crosslink actin filaments into a coherent mechanical network that allows transmission of force from one side of the cell to the other.44 In neutrophils, the actin cytoskeletal network appears highly organized and interconnected (supplemental Video 4) and thus might provide a structural framework for the putative mechanotransduction activity. Alternatively, force might be transmitted through connections among different types of cytoskeletons, as shown in other cells and organisms.41,47 Thus, MLCK inhibition at the leading edge could also affect the structure and organization of the actin cytoskeleton at the trailing edge of the cells, leading to a decrease in tractions at the rear. Similarly, this model (Figure 7) also explains the decreased tractions at both edges of the cells after p160-ROCK is inhibited.
However, it is of note that the proposed model (Figure 7) cannot explain how p160-ROCK-inhibited neutrophils retain periodicity of tractions at both edges on stiffer substrate (100 kPa). It might be that a higher-stiffness substrate helps to better support and maintain cytoskeletal organization and integrity in neutrophils, rendering cells less susceptible to local inhibition of myosin II. Indeed, the degree of integrity and organization of actin cytoskeleton are closely related to mechanical properties of the extracellular environment. More bundled, ordered stress fiber formation is observed in fibroblasts on stiffer matrix.48 Furthermore, p160-ROCK, unlike MLCK, is not required for leading edge activities and thus, when inhibited, exerts little effects on leading edge protrusion and stability. As such, the remaining myosin II activity after p160-ROCK inhibition could still enable the cells to maintain the pattern of tractions on both the leading and the trailing edges. An alternative possibility is that the putative myosin II-independent mechanism(s), as discussed in the second paragraph of this section, might be actively engaged to support the periodic pattern of tractions at the back of the cells under these conditions.
How does MLCK control integrin activation in neutrophils? Our results, as well as those from others, have begun to point to a role for myosin II-dependent mechanical tension. Friedland et al49 reported that α5β1-integrin can switch between relaxed and tensioned states in response to myosin II-generated cytoskeletal force. The resulting force combines with cell-substrate adhesion to generate tension that activates the integrin molecule mechanically. Our results also suggest that myosin II-dependent cellular contraction mediates the MLCK's effect on adhesion and integrin activation. MLCK inhibition fails to prevent integrin activation in neutrophils in suspension, implying a role of tractions in the regulation of integrin activity. We suspect that the tension applied to specific molecules (eg, talin50 or vinculin51 ) that control integrin activity might change their conformations to “active,” as in the “conformational switch” model52 proposed to explain the maturation of focal adhesions by internal actin-myosin contraction in fibroblasts. In support of this notion, application of physiologically relevant forces causes stretching of single talin rods that expose cryptic binding sites for vinculin.53 Thus, the documented protein stretching may represent a more general mechanism for force transduction in a variety of signaling events, including integrin activation.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Dr Anne Bresnick for providing the sMLCK-GFP construct, Dr Rick Horwitz for providing EGFP-α5-integrin and mCherry–myosin IIA, and the members of the F.W. laboratory for helpful discussions.
This work was supported in part by the National Institutes of Health (grant GM083812, F.W.; grant GM083601, F.W.; grant HD059002, F.W.; grant GM072744, N.W.), the Illinois Regenerative Medicine Institute (IDPH 2006-05516; F.W.), National Science Foundation (Career award 0953267; F.W.), and the University of Illinois (Arnold O. Beckman award; F.W.).
National Institutes of Health
Authorship
Contribution: M.E.S., Y.H., D.L., S.N., F.C., Y.-C.P., O.C., and P.S. performed research; P.d.L. and M.A.S. contributed vital reagents; and N.W. and F.W. designed research.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for O.C. is Functional Imaging of Transcription, UMR 8197, Institut de Biologie Ecole Normale Superieure Paris, Paris, France.
Correspondence: Fei Wang, Department of Cell and Developmental Biology, University of Illinois at Urbana-Champaign, 601 S Goodwin Ave, Urbana, IL 61801; e-mail: feiwang@life.uiuc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal