

Abstract
Following antiretroviral therapy, a significant proportion of HIV+ patients with mycobacterial coinfections develop a paradoxical, poorly understood inflammatory disease termed immune reconstitution inflammatory syndrome (IRIS). Here, we show that Mycobacterium avium–infected T cell–deficient mice injected with CD4 T cells also develop an immune reconstitution disease (IRD) manifesting as weight loss, impaired lung function, and rapid mortality. This form of IRD requires Ag recognition and interferonγ production by the donor CD4 T cells and correlates with marked alterations in blood and tissue CD11b+ myeloid cells. Interestingly, disease is associated with impaired, rather than augmented, T-cell expansion and function and is not strictly dependent on lymphopenia-induced T-cell proliferation. Instead, our findings suggest that mycobacterial-associated IRIS results from a heightened sensitivity of infected lymphopenic hosts to the detrimental effects of Ag-driven CD4 T-cell responses.
Introduction
Prolonged T-cell deficiency typically results in increased susceptibility to microbial infection, and normalization of T-cell numbers usually benefits the host through restoration of protective immunity. However, T-cell recovery after a period of lymphopenia is sometimes associated with the development of inflammatory and autoimmune disease. Currently, the most striking, prevalent manifestation of immune reconstitution disease (IRD) is observed in a subpopulation of AIDS patients during the first weeks after the initiation of antiretroviral therapy (ART). This form of IRD, referred to as immune reconstitution inflammatory syndrome (IRIS), has become a major problem in the clinical management of the HIV pandemic. Indeed, most deaths of HIV patients who receive ART occur within the first few months after the initiation of treatment,1,2 and IRIS may account for at least ∼ 10% of this early mortality.3,4
Several clinical parameters have been shown to be major predisposing factors for IRIS. These include extremely low CD4 T-cell counts (< 100 cells/μL) and the presence of preexisting or treated opportunistic infections (OIs) or malignancies at the time of ART initiation.5 In particular, IRIS is frequently observed in individuals with Mycobacterium tuberculosis (Mtb), Cryptococcus neoformans, or M avium (MAC) infections. It is generally assumed that IRIS is mediated by pathogen-specific CD4 T cells, because both MAC- and Mtb-associated reconstitution diseases are characterized by conversion to skin-test positivity to mycobacterial antigens and the appearance of granulomatous tissue pathology.6 In more recent studies of Mtb/HIV coinfected patients undergoing ART, an association has been observed between increased expansion of interferon (IFN)γ-secreting, Mtb-specific CD4 T cells and the development of IRIS symptoms.7,8 Nevertheless, this correlation between IRIS episodes and increased T helper 1 cell (Th1) response was not seen in a larger longitudinal study.9 Thus, it remains unclear if mycobacterial IRIS is due to uncontrolled T-cell expansion, if Th1 cytokines do in fact play a role, or if Ag recognition by T cells is even required.
Although IRIS is a disease of lymphopenic patients, it is not clear what factors in T cell–deficient hosts drive destructive CD4 T-cell responses during immune reconstitution. One hypothesis is that lymphopenia itself may lead to T-cell dysregulation. Indeed, it is well established that in the absence of cognate Ag, resting T cells spontaneously proliferate and acquire activation markers when introduced into T cell–deficient hosts.10 Ag-driven T-cell responses are also dramatically dysregulated in T-cell lymphopenic environments. This is, perhaps, best illustrated by the breakdown of peripheral T-cell tolerance in lymphopenic conditions.11 Multiple studies in mice have demonstrated a role for lymphopenia-induced T-cell activation in the development of autoimmunity.12-19 In humans, it has been shown that T-cell depletion with alemtuzumab (a humanized anti-CD52 monoclonal antibody [mAb]) can lead to the development of autoimmune disease.20-22 In cancer patients undergoing T-cell immunotherapy, lymphodepletion before T-cell transfer greatly increases the antitumor function of the donor cells,23 and, in mice, lymphopenia can reverse tumor-specific T-cell anergy.24 On the basis of such observations, it was proposed that lymphopenia-induced T-cell activation might be an important factor in the pathogenesis of IRIS.25 Nevertheless, a role for this mechanism has never been formally established.
Many of the above mechanistic issues in the understanding of IRIS development are difficult to formally address in clinical studies. For this reason, we considered it important to develop an experimental animal model of IRIS that recapitulates the basic immunologic scenario of the human disease and that can be used to understand the fundamental principles of CD4 T-cell repopulation of lymphopenic hosts in the presence of mycobacterial infection. To perform this analysis, we used T cell–deficient T-cell receptor (TCR)α−/− mice infected with M avium, an important opportunistic pathogen associated with human IRIS.26 At moderate infectious doses, M avium infection in T cell–deficient mice is asymptomatic for many months, despite disseminated bacterial growth.27 Nevertheless, as demonstrated here, these animals develop a severe wasting disease and rapidly succumb when adoptively transferred with CD4 T cells. We have used this experimental model to examine the requirements for T-cell expansion, the role of Th1 cytokines, and the requirement for Ag recognition in the immunopathogenesis of IRD. Unexpectedly, our findings reveal that reconstitution disease in the setting of mycobacterial infection is associated with impaired, rather than augmented, T-cell expansion and, instead, appears to reflect the heightened sensitivity of infected lymphopenic hosts to the deleterious effects of reemerging Ag-driven Th1 responses.
Methods
Mice
C57BL/6 were purchased from Taconic Farms. TCRα−/−, B6.SJL, IFNγ−/−, interleukin (IL)-4−/−, Foxp3eGFP reporter mice, OT-II, and the male-Ag–specific Marilyn TCR Tg mice were obtained through a special contract between the National Institute of Allergy and Infectious Diseases (NIAID) and Taconic Farms. The Ag8b-specific P25 TCR Tg mice were originally produced by Takatsu et al28 and were generously provided by Dr Joel Ernst. IL-17A−/− mice29 were generously provided by Dr Rachel Caspi. Enhanced yellow fluorescent protein (eYFP) IFNγ YETI reporter mice were originally derived by Richard Locksley and colleagues30 and are maintained in our laboratory. All animals were bred and housed at an AAALAC-approved facility at the NIAID/NIH (National Institutes of Health, Bethesda, MD), according to the Guide for the Care and Use of Laboratory Animals. Mice were used according to an animal study proposal approved by the NIAID Animal Care and Use Committee.
Mycobacterial infections and measurement of bacterial loads
Mice (6-8 weeks old) were innoculated intravenously with 1 × 106 colony-forming units of M avium (strain SmT 2151). Bacterial loads in infected mice were quantified by plating tissue homogenates on oleic acid-albumin-dextrose-catalase (OADC)–supplemented Middlebrook 7H11 agar (Difco) and counting colonies on day 14 of incubation.
Cell purifications and adoptive transfers
CD4 T cells for adoptive transfer were enriched from the spleen and lymph nodes of uninfected mice by MACS magnetic beads (Miltenyi Biotec) to approximately 90%-95% purity, according to the manufacturer's protocol. In the experiments using IFNγ and Foxp3 reporter mice, CD4 T cells were first enriched using anti-CD4 MACS beads and then further flow cytometrically fractionated using a Becton Dickinson FACSVantage SE or FACSAria to > 95% purity. Where indicated, enriched CD4 T cells were stained with 1μM carboxyfluorescein succinimidyl ester (CFSE) at 1 × 106 cells/mL for 10 minutes at room temperature before adoptive transfer (Molecular Probes-Invitrogen).
Pulse oximetry
Percent O2 saturation was measured using the MouseOx small-animal pulse oximeter from STARR Life Sciences Corp. Mice were shaved, and a collar sensor was outfitted to the back of the neck. Measurements were made on unanesthetized, unrestrained mice using the manufacturer's instrument software and represent the average over 15 seconds of stable monitoring.
Cytokine ELISA and nitric oxide quantification
Serum IFNγ and tumor necrosis factor α (TNFα) were measured using enzyme-linked immunosorbent assay (ELISA) kits from eBioscience and R&D DuoSet ELISA kits, respectively. Serum nitric oxide was measured using the Total NO/nitrite/nitrate kit from R&D Systems.
Antibodies and flow cytometry
Anti-CD4 (RM4-4), CD45.1 (A20), Thy1.1 (H1S51), TCRβ (H57-597), IFNγ (XMG1.2), CD44 (IM7), CD11b (M1/70), CD115 (AFS98), CD11c (HL3), Ly6C (HK1.4), Gr-1 (RB6-8C5), and CD69 (H1.2F3) antibodies were purchased from eBioscience, Biolegend, and BD Pharmingen. Biotinylated antibodies were detected with streptavidin-conjugated Qdot 605 from Molecular Probes-Invitrogen. Annexin V Pacific Blue and ultraviolet (UV) and near infrared (IR) fixable live/dead cell stains were obtained from Molecular Probes-Invitrogen and used according to the manufacturer's protocol. All samples were acquired on a LSRII flow cytometer (Becton Dickinson).
In vivo TNFα blockade
Mice were injected intraperitoneally with 500 μg of anti-TNFα (XT3.11) or irrelevant rat immunoglobulin G1 (IgG1) isotype control (HRPN) from BioXCell every 3 days after T-cell transfer.
Weight-loss calculation and statistical analysis
Weight loss, determined as an index of wasting, was calculated as the percentage change from initial weight and was normalized to the weight taken immediately before transfer of T cells:
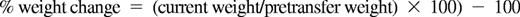
The statistical significance of differences between data groups was analyzed by Student t test using GraphPad Prism software (Version 5.0c). P was considered statistically significant at a value of P < .05 or less.
Results
Transfer of CD4 T lymphocytes into T cell–deficient M avium–infected mice results in a fatal wasting disease
To examine the outcome of T-cell reconstitution of lymphopenic hosts during chronic mycobacterial infection, we transferred 2 × 106 purified CD4 T cells from naive donors into T cell–deficient TCRα−/− mice that had been infected with M avium 2-3 months previously (Figure 1A). These recipients underwent rapid wasting, losing 20%-30% of their initial weight between days 6 and 10 after transfer, and all succumbed by day 21 (Figure 1B). In contrast, infected C57Bl/6 wild-type (WT) recipients of the same number of CD4 T cells displayed no significant weight loss or mortality, nor, as expected, did T cell–reconstituted, uninfected TCRα−/− recipients (data not shown). Animals reconstituted with T cells had similar bacterial loads as unreconstituted TCRα−/− mice 9 days after transfer (Figure 1C), indicating that the disease was not due to an effect on pathogen control. Although widely disseminating into most tissues, M avium preferentially replicates in the lungs of mice. Using a pulse oximeter designed for rodents, we found that while infected TCRα−/− mice had O2 saturation levels ∼ 90% before transfer, by 8 days after transfer, CD4 T-cell recipients had decreased to ∼ 65% O2 saturation (Figure 1D). These findings demonstrate that in the presence of concurrent T-cell deficiency and mycobacterial infection, CD4 T-cell transfer is sufficient to drive a severe immune reconstitution disease that results in wasting, impaired lung function, and death.

Immune reconstitution disease in Mycobacterium avium–infected T cell–deficient mice after CD4 T-cell adoptive transfer. (A) TCRα−/− mice were infected intravenously with 1 × 106 colony-forming units of M avium. After at least 2 months, 2 × 106 CD4 T cells isolated from uninfected WT mice were transferred intravenously. (B) Mice were monitored for weight loss (n > 11 mice/group) and survival (n = 8 mice/group). The shaded area between traveling error bars represents the SD. (C) Bacterial loads were measured in the lungs and spleens of control and T cell–recipient TCRα−/− mice on day 9 of transfer. (D) Lung function was assessed by measuring percent O2 saturation using a murine pulse oximeter on day 8 after transfer. Data are representative of at least 2 independent experiments performed, and the results shown in panel B have been repeated at least 15 times.
Immune reconstitution disease in Mycobacterium avium–infected T cell–deficient mice after CD4 T-cell adoptive transfer. (A) TCRα−/− mice were infected intravenously with 1 × 106 colony-forming units of M avium. After at least 2 months, 2 × 106 CD4 T cells isolated from uninfected WT mice were transferred intravenously. (B) Mice were monitored for weight loss (n > 11 mice/group) and survival (n = 8 mice/group). The shaded area between traveling error bars represents the SD. (C) Bacterial loads were measured in the lungs and spleens of control and T cell–recipient TCRα−/− mice on day 9 of transfer. (D) Lung function was assessed by measuring percent O2 saturation using a murine pulse oximeter on day 8 after transfer. Data are representative of at least 2 independent experiments performed, and the results shown in panel B have been repeated at least 15 times.
Differential expansion of donor CD4 T cells in naive and M avium–infected T cell–deficient mice
It is well known that CD4 T cells spontaneously proliferate when introduced into T cell–deficient environments, but the effect of bacterial infection on the “emptiness-driven” proliferation of CD4 T cells is not well understood. One hypothesis to explain mycobacterial-associated IRD is that concurrent bacterial infection results in exaggerated CD4 T-cell expansion and activation after ART. To examine this question, we compared the expansion in number and proliferation of CFSE-labeled CD45.1+ donor CD4 T cells in infected versus uninfected CD45.2+ TCRα−/− recipient mice. Interestingly, in each tissue examined, there were approximately 2- to 5-fold fewer donor cells recovered from infected compared with naive recipients (Figure 2A-B). To ask whether the poor recovery of T cells from the infected mice was due to a lack of cell division, we next examined CFSE dilution. When compared on day 7 after transfer, similar percentages (> 90%) of donor cells had completely diluted their CFSE in infected and uninfected recipients as measured in the spleen, liver, lung, bone marrow, and mediastinal lymph node (Figure 2A). In the peripheral blood mononuclear cells (PBMCs), however, there was a slight reduction in the frequency of donor cells that had divided in the infected recipient mice (97% vs 68% CFSE). Nevertheless, while recovered in lower numbers, the donor T cells in the PBMCs, spleen, liver, lungs, and bone marrow of M avium–infected mice expressed much higher levels of the activation marker, CD69, than their counterparts in naive recipients (Figure 2C). These data indicate that the CD4 T cells that trigger disease in M avium–infected mice, although highly activated and undergoing cell division in most tissues, fail to accumulate in the same numbers as in uninfected recipients, which do not develop disease.

Donor CD4 T-cell expansion and activation during reconstitution of naive and M avium–infected TCRα−/− mice. Naive and day 69 infected CD45.2+ TCRα−/− mice received 1.5 × 106 CFSE-labeled CD45.1+ CD4 T cells from naive donors, and tissues were harvested on day 7 after transfer. (A) Representative FACS plots showing the frequency of donor CD4 T cells in different tissues of naive and infected recipient mice (top panels). Numbers represent the percentage of the total recovered cells that were of donor origin. The corresponding histograms of CFSE dilution for each of the gated CD4+CD45.1+ donor cell populations are shown in the bottom panels. Numbers represent the percentage of donor cells that have fully diluted CFSE. (B) Absolute numbers of donor CD4 T cells recovered from each of the tissues shown in (A). For the PBMCs, the percentage of donor cells is shown. (C) CD69 staining of the donor CD4 T cells recovered in panels A and B. In the case of mediastinal lymph nodes, it was necessary to pool cells from 4 mice to obtain sufficient numbers for staining. Data are representative of 2 independent experiments.
Donor CD4 T-cell expansion and activation during reconstitution of naive and M avium–infected TCRα−/− mice. Naive and day 69 infected CD45.2+ TCRα−/− mice received 1.5 × 106 CFSE-labeled CD45.1+ CD4 T cells from naive donors, and tissues were harvested on day 7 after transfer. (A) Representative FACS plots showing the frequency of donor CD4 T cells in different tissues of naive and infected recipient mice (top panels). Numbers represent the percentage of the total recovered cells that were of donor origin. The corresponding histograms of CFSE dilution for each of the gated CD4+CD45.1+ donor cell populations are shown in the bottom panels. Numbers represent the percentage of donor cells that have fully diluted CFSE. (B) Absolute numbers of donor CD4 T cells recovered from each of the tissues shown in (A). For the PBMCs, the percentage of donor cells is shown. (C) CD69 staining of the donor CD4 T cells recovered in panels A and B. In the case of mediastinal lymph nodes, it was necessary to pool cells from 4 mice to obtain sufficient numbers for staining. Data are representative of 2 independent experiments.
Major role for Th1 cytokines in reconstitution disease
To examine the role of T cell–derived cytokines in our immune reconstitution disease model, we transferred WT, IFNγ−/−, IL-4−/−, or IL-17A−/− CD4 T cells into M avium–infected TCRα−/− mice and monitored their weight loss and survival. Recipients of WT, IL-4−/−, and IL-17A−/− CD4 T cells displayed the same rapid weight loss, and all succumbed by day 24 (Figure 3A-B). In contrast, in recipients of IFNγ−/− CD4 T cells, both weight loss and mortality were delayed and death of all the animals was not observed until day 57 after transfer (Figure 3A-B). Significantly, when TCRβ+CD4+ cells were quantified in the blood of the recipients on day 9, the IFNγ−/− donor CD4 T cells were found to have expanded to approximately 5× the level of WT, IL-4−/−, or IL-17A−/− T cells (Figure 3C). These findings confirm that the attenuated disease in the recipients of IFNγ−/− CD4 T cells is not due to a defect in donor-cell reconstitution.

CD4 T cell–derived IFNγ, but not IL-4 or IL-17A, plays a major role in immune reconstitution disease of M avium–infected TCRα−/− mice. Totals of 2 × 106 WT, IL-4−/−, IL-17A−/−, or IFNγ−/− CD4 T cells were transferred into M avium–infected TCRα−/− mice (n = 5 mice/group), as described in Figure 1, and (A) weight loss and (B) survival were monitored. The data shown in (A) are representative of 2-3 independent experiments, while the survival curves shown in (B) represent 9 mice pooled from 2 independent experiments. (C) TCRβ+CD4+ cells were measured in the PBMCs of the mice shown in panels A and B on day 9 after transfer. (D) IFNγ was measured by ELISA in the serum of the same animals on day 10 after transfer. (E) Levels of nitrate were measured in the same sera using a nitrate reductase assay, and TNFα levels (F) were determined by ELISA. (G) M avium–infected TCRα−/− mice were reconstituted with WT CD4 T cells and then treated with anti-TNFα–neutralizing Ab or isotype control Ab on days 0, 3, 6, 9, and 12 after transfer, and weight loss was monitored. The data shown for each panel are representative of at least 2 independent experiments. In panels A and G, the shaded area between the traveling error bars represents the SD.
CD4 T cell–derived IFNγ, but not IL-4 or IL-17A, plays a major role in immune reconstitution disease of M avium–infected TCRα−/− mice. Totals of 2 × 106 WT, IL-4−/−, IL-17A−/−, or IFNγ−/− CD4 T cells were transferred into M avium–infected TCRα−/− mice (n = 5 mice/group), as described in Figure 1, and (A) weight loss and (B) survival were monitored. The data shown in (A) are representative of 2-3 independent experiments, while the survival curves shown in (B) represent 9 mice pooled from 2 independent experiments. (C) TCRβ+CD4+ cells were measured in the PBMCs of the mice shown in panels A and B on day 9 after transfer. (D) IFNγ was measured by ELISA in the serum of the same animals on day 10 after transfer. (E) Levels of nitrate were measured in the same sera using a nitrate reductase assay, and TNFα levels (F) were determined by ELISA. (G) M avium–infected TCRα−/− mice were reconstituted with WT CD4 T cells and then treated with anti-TNFα–neutralizing Ab or isotype control Ab on days 0, 3, 6, 9, and 12 after transfer, and weight loss was monitored. The data shown for each panel are representative of at least 2 independent experiments. In panels A and G, the shaded area between the traveling error bars represents the SD.
Consistent with the above observations, marked increases in IFNγ were detected in sera of M avium–infected recipients of WT, but not IFNγ−/− CD4 T cells (Figure 3D). IFNγ is known to play a major role in the production of nitric oxide by inducing the expression of NOS2, and, accordingly, we found that transfer of WT T cells, but not IFNγ−/− T cells, led to elevated levels of nitrate in the serum (Figure 3E). Nitric oxide has previously been demonstrated to be a major inhibitor of T-cell expansion in vivo during M avium infection,31 and thus the impaired NO production seen in the recipients of IFNγ−/− T cells therefore is likely to explain the increased expansion of donor cells in these animals.
We also observed a reduced induction of TNFα in the serum of recipients of IFNγ−/−, compared with WT CD4 T cells (Figure 3F), so we considered the possibility that the principle role of IFNγ in this reconstitution disease is to trigger the production of the former cytokine. To test this hypothesis, M avium–infected TCRα−/− mice receiving WT CD4 T cells were treated with anti-TNFα–neutralizing mAb. As a consequence of TNFα blockade, these animals, while initially gaining weight, starting on day 5, began to rapidly waste and were euthanized by day 14 (Figure 3G). Interestingly, when quantified on day 9 after transfer, the expansion of the CD45.1+ donor CD4 T cells was found to be markedly increased in the recipients of anti-TNFα versus isotype control Ab (5.5% vs 0.3% of the total PBMC, data not shown). Collectively, the above experiments demonstrated that IFNγ and, to a lesser extent, TNFα are critical mediators of both the immune reconstitution disease occurring in M avium–infected TCRα−/− mice and, ironically, the suppression of lymphocyte expansion seen in these animals.
The disease inducing CD4 T cells derive largely from precursors with a naive phenotype
We next examined the phenotype of the CD4 T cells in normal spleen and lymph nodes (LNs) responsible for driving reconstitution disease. Since IFNγ-producing CD4 T cells were shown above (Figure 3) to play a major role in this model, we next asked if the Th1 cells preexisting in naive mice are the precursors of the pathogenic cells. To do so, we used IFNγ reporter (YETI) mice, which express enhanced yellow fluorescent protein under control of the IFNγ promoter. CD4 T cells from naive CD45.1+ congenic YETI mice were flow sorted into eYFP+ (IFNγ expressing) and eYFP− (IFNγ negative) CD4 T-cell populations and transferred into CD45.2+M avium–infected TCRα−/− mice (Figure 4A). Mice receiving eYFP− CD4 T cells lost 25% of their body weight by day 13, while recipients of eYFP+ CD4 T cells displayed no signs of illness (Figure 4A). When examined on day 10 after transfer, 1% of the PBMCs in mice receiving eYFP− cells were donor derived, and of these, 7% now expressed eYFP, indicating that a subpopulation of these cells had differentiated into Th1 cells after adoptive transfer. In contrast, in the mice receiving eYPF+ CD4 T cells, 0.01% of the PBMCs were of donor origin and only 16% of the remaining cells still expressed eYFP (Figure 4A). These data demonstrate that the precursors of the pathogenic cells in the bulk CD4 T-cell inocula are initially negative for IFNγ message and up-regulate its expression after transfer.

The naive T-cell subpopulation is largely responsible for the induction of immune reconstitution disease by bulk CD4 T cells. (A) CD4 T cells from uninfected IFNγ reporter mice were FACS sorted into eYFP+ and eYFP− cells. Purified cells (5 × 105) of each subset were then separately transferred into M avium–infected TCRα−/− mice (n = 6 control mice, n = 5 recipients of eYFP−, and n = 4 recipients of eYFP+ CD4 T cells). On day 10 after transfer, the recipient mice were analyzed for the donor cell marker and eYFP expression. (B) CD4 T cells from foxp3 reporter mice were FACS purified into CD44lofoxp3−, CD44hifoxp3−, and foxp3+ cells. Purified cells (5 × 105) of each subset were then separately transferred into M avium–infected TCRα−/− mice, and recipients were monitored for weight loss and survival (n = 5: recipients of CD44lofoxp3− or CD44hifoxp3− cells and n = 3: recipients of foxp3+ T cells). In panels A and B, the shaded area between the traveling error bars represents the SD. All data are representative of 2 independent experiments performed.
The naive T-cell subpopulation is largely responsible for the induction of immune reconstitution disease by bulk CD4 T cells. (A) CD4 T cells from uninfected IFNγ reporter mice were FACS sorted into eYFP+ and eYFP− cells. Purified cells (5 × 105) of each subset were then separately transferred into M avium–infected TCRα−/− mice (n = 6 control mice, n = 5 recipients of eYFP−, and n = 4 recipients of eYFP+ CD4 T cells). On day 10 after transfer, the recipient mice were analyzed for the donor cell marker and eYFP expression. (B) CD4 T cells from foxp3 reporter mice were FACS purified into CD44lofoxp3−, CD44hifoxp3−, and foxp3+ cells. Purified cells (5 × 105) of each subset were then separately transferred into M avium–infected TCRα−/− mice, and recipients were monitored for weight loss and survival (n = 5: recipients of CD44lofoxp3− or CD44hifoxp3− cells and n = 3: recipients of foxp3+ T cells). In panels A and B, the shaded area between the traveling error bars represents the SD. All data are representative of 2 independent experiments performed.
We then asked whether disease induction is mediated preferentially by naive, Treg, or effector/memory cells preexisting in the donor population. CD4 T cells from foxp3 reporter mice were FACS purified into 3 populations: foxp3+ regulatory T cells; CD44highfoxp3− effector/memory T cells; and CD44lowfoxp3− naive T cells. Although recent reports have demonstrated that, under certain conditions, regulatory T cells can lose foxp3 expression and become pathogenic effector cells,32,33 we observed no disease after transfer of purified foxp3+ CD4 T cells, indicating that deprogrammed Tregs are unlikely to be pathogenic in our model. Mice receiving CD44highfoxp3− CD4 T cells lost ∼17% of their body weight by day 12 after transfer, and all were still alive 40 days after transfer (Figure 4B). In contrast, mice receiving CD44lowfoxp3− CD4 T cells lost > 25% of their body weight by day 12, and 80% of the mice succumbed before day 30 (Figure 4B). These data indicated that CD4 T lymphocytes with a naive phenotype are the major source of the disease-inducing precursors, although cells with an effector/memory phenotype preexisting in naive mice can contribute to a lesser extent.
Recognition of bacterial or endogenously expressed Ag by CD4 T cells leads to disease induction
To test if Ag recognition is required to drive reconstitution disease, we transferred TCR Tg CD4 T cells that recognize an irrelevant Ag (ovalbumin-specific OT-II CD4 T cells) or M avium–specific TCR Tg CD4 T cells (P25 T cells recognizing Ag85b) into M avium–infected TCRα−/− mice. Adoptive transfer of OT-II cells induced no weight loss or mortality (Figure 5A), while infected, but not uninfected, recipients of P25 T cells displayed aggressive disease progression, with 100% of the animals succumbing by day 8 (Figure 5B). We also measured the expansion in number, division, and death of the P25 T cells. On day 4 after transfer, we recovered approximately 4-fold fewer donor cells from infected, compared with naive, recipients, consistent with the decreased recovery of polyclonal donor CD4 T cells shown in Figure 2. When CFSE dilution was analyzed, we found that the transferred P25 T cells had not yet divided in naive recipients, but had extensively diluted their CFSE in infected recipients (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Analysis of annexin V and a dead cell–specific dye demonstrated that the failure of the dividing cells to accumulate in the infected mice was likely due to a very high rate of cell death (supplemental Figure 1). Because no alterations in bacterial burden were observed in P25 recipients, their wasting and mortality could not be explained by changes in pathogen load (Figure 5C). Together, these findings argued that the ability to induce IRD is not a fundamental property of all naive T cells, and that Ag recognition by CD4 T cells is required for disease induction. Furthermore, the data demonstrated that the mycobacterial infection itself can serve as the target of the pathogenic T-cell response.

CD4 T cells specific for M avium can drive immune reconstitution disease. (A) M avium–infected TCRα−/− mice received CD4 T cells (2 × 106 each) isolated from uninfected WT or OT-II mice. The shaded area between the traveling error bars represents the SD. (B) Uninfected or M avium–infected TCRα−/− mice received P25 (Ag85b specific TCR Tg) CD4 T cells (2 × 106) or no T cells, and survival was monitored. (C) Bacterial loads in the lungs of infected mice from panel B 7 days after transfer of P25 CD4 T cells. (D) Total nitrate and (E) TNFα were measured in the serum of mice from panel B on day 5 of P25 transfer. (F) CD11b+ cells were measured in the blood and (G) lung of mice from panel B on day 5 of P25 transfer. (H) Phenotype of CD11b+ cells in the PBMC of M avium–infected TCRα−/− mice on day 6 of P25 transfer.
CD4 T cells specific for M avium can drive immune reconstitution disease. (A) M avium–infected TCRα−/− mice received CD4 T cells (2 × 106 each) isolated from uninfected WT or OT-II mice. The shaded area between the traveling error bars represents the SD. (B) Uninfected or M avium–infected TCRα−/− mice received P25 (Ag85b specific TCR Tg) CD4 T cells (2 × 106) or no T cells, and survival was monitored. (C) Bacterial loads in the lungs of infected mice from panel B 7 days after transfer of P25 CD4 T cells. (D) Total nitrate and (E) TNFα were measured in the serum of mice from panel B on day 5 of P25 transfer. (F) CD11b+ cells were measured in the blood and (G) lung of mice from panel B on day 5 of P25 transfer. (H) Phenotype of CD11b+ cells in the PBMC of M avium–infected TCRα−/− mice on day 6 of P25 transfer.
As was observed previously with transferred bulk CD4 T cells, infected recipients of mycobacteria-specific P25 CD4 T cells showed marked elevations in serum total nitrate and TNFα (Figure 5D-E). These findings led us to consider whether the IRD seen in our model is associated with alterations in the myeloid cell compartment. On day 5 after transfer, infected, but not naive, recipients of P25 CD4 T cells displayed increased frequencies of CD11b+ cells in both the peripheral blood (Figure 5F) and lung (Figure 5G). Further characterization of this CD11b+ population in blood revealed an increase in CD11b+Ly6CintGr-1hiCD115low and CD11b+Ly6ChiCD115hi cells accompanied by a simultaneous decrease in the CD11b+Ly6clowGr-1negCD115+CD11c+ cells when measured on day 6 after T-cell transfer (Figure 5F and data not shown). Based on their phenotypic markers, the increased populations are likely to represent inflammatory monocytes and neutrophils.
We next asked if bacteria-specific CD4 T cells are required to drive disease by adoptively transferring TCR Tg CD4 T cells that recognize a host tissue Ag. To do this, we transferred T cells specific for an Ag present only in male mice (Marilyn CD4 cells) into naive or infected female or male TCRα−/− mice (Figure 6A). As expected, naive or infected female recipients of Marilyn CD4 T cells failed to display reconstitution disease (Figure 6B), further demonstrating that Ag recognition by CD4 T cells is required for pathology. Similarly, no disease was observed in uninfected naive males receiving Marilyn CD4 T cells (Figure 6B). In direct contrast, 100% of the M avium–infected male recipients underwent wasting and succumbed by day 6 after transfer (Figure 6B). A similar outcome was seen in a parallel model using the transfer of pigeon cytochrome c (PCC)–specific TCR Tg CD4 T cells into T cell–deficient PCC-expressing recipients (data not shown). Taken together with the previous data on OT-II and P25 transfer, these experiments argued that disease induction requires Ag recognition, but that the Ag recognized need not be of bacterial origin.

CD4 T cells specific for endogenous antigens can drive immune reconstitution disease. (A) M avium–infected and –uninfected male or female TCRα−/− mice received Marilyn (antimale TCR Tg) CD4 T cells (2 × 106) isolated from female donor mice. (B) Survival of mice in shown in panel A. (C) IFNγ and (D) total nitrate were measured in the serum of mice shown in panel A on day 3 after transfer. All data are representative of at least 2 independent experiments.
CD4 T cells specific for endogenous antigens can drive immune reconstitution disease. (A) M avium–infected and –uninfected male or female TCRα−/− mice received Marilyn (antimale TCR Tg) CD4 T cells (2 × 106) isolated from female donor mice. (B) Survival of mice in shown in panel A. (C) IFNγ and (D) total nitrate were measured in the serum of mice shown in panel A on day 3 after transfer. All data are representative of at least 2 independent experiments.
Importantly, after transfer of anti-male CD4 T cells, we observed lower levels of IFNγ in the serum of infected, compared with uninfected, males, despite their increased mortality (Figure 6C). This finding suggested that the former recipients are more susceptible to the deleterious effects of IFNγ than are the latter. Consistent with this hypothesis, we found that infected male recipients of Marilyn CD4 T cells displayed increased production of nitric oxide, compared with naive male recipients (Figure 6D). Thus, in this system, where the T cells are not directed against the mycobacteria themselves, these data indicate that mycobacterial infection appears to promote disease by making the host more responsive to the cytokines produced by the reconstituting T cells.
T cell–induced reconstitution disease does not require a lymphopenic environment
An important hypothesis is that in HIV+ individuals, CD4 T-cell deficiency leads to susceptibility to IRD due to the lymphopenia-induced T-cell expansion and activation that occurs after ART. However, it is also possible that susceptibility of infected lymphopenic hosts stems, instead, from the deficiency in Ag-specific CD4 T cells recognizing the opportunistic pathogen. We reasoned that these 2 hypotheses could be distinguished in our model by examining the effect on disease induction of general lymphopenia versus the specific inability to recognize and respond to M avium. To do so, we first compared the outcome of the transfer of CFSE-labeled Thy1.1+ P25 CD4 T cells into uninfected nonlymphopenic Thy1.2+ B6 mice, lymphopenic TCRα−/− mice, and OT-II recipients. The latter animals provide a nonlymphopenic setting in which the mice are unable to respond to M avium. As expected, when measured in the LN on day 7 after transfer, extensive donor-cell proliferation was evident in TCRα−/− mice, but not in B6 recipients (Figure 7A). Importantly, a lack of donor cell expansion was also seen in the OT-II recipients, indicating that the P25 CD4 T cells do not sense either the B6 or OT-II recipient mice as lymphopenic.

Immune reconstitution disease is associated with the prior absence of host M avium–specific T-cell responses, rather than lymphopenia-driven T-cell expansion. (A) Uninfected WT, TCRα−/−, and OT-II recipients received CFSE-labeled Thy1.1+ P25 CD4 T cells (2 × 106/mouse), and lymph nodes were harvested on day 7 after transfer and analyzed for CFSE dilution. Histograms are gated on CD4+Thy1.1+ donor P25 CD4 T cells. (B) M avium–infected WT, TCRα−/−, or OT-II mice were injected with P25 CD4 T cells (2 × 106), and survival was monitored. (C) CD4 T cells (2 × 106) isolated from C57Bl/6 mice were transferred into M avium–infected TCRα−/−, OT-II, or P25 recipient mice and weight loss was monitored. The shaded area between the traveling error bars represents the SD. (D) Survival of mice depicted in panel C, n = 5/group. All of the data shown were representative of 2 independent experiments performed.
Immune reconstitution disease is associated with the prior absence of host M avium–specific T-cell responses, rather than lymphopenia-driven T-cell expansion. (A) Uninfected WT, TCRα−/−, and OT-II recipients received CFSE-labeled Thy1.1+ P25 CD4 T cells (2 × 106/mouse), and lymph nodes were harvested on day 7 after transfer and analyzed for CFSE dilution. Histograms are gated on CD4+Thy1.1+ donor P25 CD4 T cells. (B) M avium–infected WT, TCRα−/−, or OT-II mice were injected with P25 CD4 T cells (2 × 106), and survival was monitored. (C) CD4 T cells (2 × 106) isolated from C57Bl/6 mice were transferred into M avium–infected TCRα−/−, OT-II, or P25 recipient mice and weight loss was monitored. The shaded area between the traveling error bars represents the SD. (D) Survival of mice depicted in panel C, n = 5/group. All of the data shown were representative of 2 independent experiments performed.
We next asked if M avium–infected OT-II TCR Tg mice are susceptible to reconstitution disease induced by M avium–specific P25 CD4 T cells. Surprisingly, these animals rapidly succumbed, with a kinetics indistinguishable from that of TCRα−/− recipients of the same donor cells, while intact B6 recipients survived, as expected (Figure 7A). The above findings indicated that a lymphopenic environment is not a prerequisite for disease induction.
We reasoned that the key difference between the disease-susceptible OT-II and the disease-resistant WT recipients is the inability of the OT-II mice to mount T-cell responses to the bacterial infection. To test this hypothesis, we next asked if recipient mice bearing only T cells specific for M avium were susceptible to reconstitution disease. To perform this experiment, we transferred B6 CD4 T cells into infected P25 recipients. The latter animals failed to develop significant wasting disease or mortality, in contrast to TCRα−/− or OT-II recipients (Figure 7C-D). Together, these findings suggested that the absence of a preexisting Ag-specific CD4 T-cell response, in this case against M avium, may be the key factor determining disease susceptibility of lymphopenic hosts undergoing T-cell reconstitution.
Discussion
In this study, we have described a new model of reconstitution disease that recapitulates the fundamental immunologic scenario of mycobacterial-associated IRIS: CD4 T-cell reconstitution of lymphopenic hosts previously exposed to mycobacteria. In this system, adoptive transfer of purified CD4 T cell leads to disease, strongly supporting the hypothesis that IRIS in mycobacterial-infected HIV patients is mediated by CD4 T cells that repopulate the host after ART. Our data also suggest that susceptibility to IRD may be a common property of microbial-infected immunodeficient hosts undergoing T-cell reconstitution. Indeed, the induction of inflammatory disease has also been described after adoptive transfer of CD4 T cells into Pneumocystis carinii–exposed scid mice.34
In the M avium IRIS model studied here, Ag recognition by T cells is required to drive disease, and bacteria-specific T cells were highly pathogenic, arguing in favor of the hypothesis that mycobacterial IRIS in patients is driven by bacteria-specific T cells. Interestingly, in some patients, IRIS displays autoimmune-like features, suggesting the involvement of self-reactive T cells. In our model, T cells recognizing Ag expressed transgenically in the infected host were also capable of disease induction. One interpretation of these findings is that in patients with IRIS, the major coinfection may serve as the trigger, rather than the target, of the pathogenic T-cell response, which, in some individuals, could be directed against self-Ag or unrelated foreign Ag (eg, from another pathogen or commensal) present in the host.
While CD4 T-cell transfer into M avium–infected TCRα−/− mice triggers disease, no IRIS-like symptoms are seen in T cell–replete WT mice. This observation is consistent with the hypothesis drawn from previous studies that T cells become spontaneously activated and their effector functions are enhanced when expanding in a lymphopenic environment. Accordingly, one hypothesis to explain IRIS is that lymphopenia-induced T-cell activation promotes uncontrolled T-cell expansion and effector function that leads to tissue destruction. However, while lymphopenic hosts support emptiness-driven T-cell proliferation and activation, because of their immunodeficiency, they also fail to mount normal pathogen-specific immune responses, and it is possible that the resulting lack of preexisting responsiveness to the infection may, itself, contribute to disease susceptibility. M avium infection of OT-II mice provides a recipient host that does not support lymphopenia-driven activation and does not mount normal responses against the mycobacterial infection. Unexpectedly, we found that M avium–infected OT-II mice were highly susceptible to reconstitution disease. Furthermore, transfer of extremely large numbers of naive CD4 T cells (108), rather than the usual smaller number (106), only accelerated mortality (data not shown). Therefore, reconstituting CD4 T cells do not need to sense a lymphopenic environment to mediate IRD in our model. At the same time, we found that infected Ag85b TCR Tg mice are largely resistant to disease, indicating that the presence of preexisting bacteria-specific CD4 T cells is sufficient to protect mice from IRD.
The above data are consistent with our observations that T-cell expansion in M avium–infected hosts is actually impaired, compared with reconstitution in naive T cell–deficient recipients. Furthermore, when CD4 T cells were isolated from naive and infected TCRα−/− mice on day 7 of transfer, we found that fewer of the donor T cells recovered from infected, compared with naive, recipients produced IFNγ upon restimulation (data not shown). Upon transfer of anti-male CD4 T cells, we also found significantly less IFNγ in the serum of M avium–infected, compared with naive male recipients, although the infected male mice rapidly succumbed, while the naive mice survived. Therefore, reconstitution disease in our model did not appear to be associated with exacerbated T-cell expansion and cytokine production. Instead, our data collectively argue that reconstitution disease is due to the increased sensitivity of infected hosts to the detrimental effects of CD4 T cell–produced IFNγ. We speculate that this augmented susceptibility results from the absence of M avium–specific CD4 T-cell responses before reconstitution in the chronically infected TCRα−/− hosts.
Ag-specific Th1 cells are potent activators of macrophage through the production of IFNγ, and M avium–infected T cell–deficient mice harbor large numbers of bacilli-containing macrophage that have been primed by microbial signals, but have never received T-cell help. One possibility is that reconstitution disease, in this setting, results from the triggering of these primed infected macrophages by lymphokines produced by the repopulating Th1 cells. Indeed, we observed the rapid induction of serum nitric oxide after transfer of WT CD4 T cells (but not IFNγ−/− CD4 T cells) into infected TCRα−/− recipients to be consistent with the occurrence of de novo macrophage activation in the former animals. CD4 T cells have also been shown to trigger the accelerated production of monocytes and neutrophils in the bone marrow after M avium infection,35 and, indeed, we observed large increases in CD11b+ cells in the blood and lungs of M avium–infected TCRα−/− mice after T-cell transfer. Moreover, our attempts to trigger reconstitution disease in M avium–infected, T cell–sufficient mice by transiently inducing CD4 T-cell depletion by treatment with mAb were unsuccessful, suggesting that the pathologic response depends on the prolonged absence of activation signals from Ag-specific T-cell help. Together, these observations suggest that the prior lack of pathogen-specific T-cell responses in infected lymphopenic hosts promotes dysregulated myeloid activation and/or myelopoesis driven by Ag-specific CD4 T cells during immune reconstitution. A role for dysregulated myeloid cell responses in IRD has also been suggested from the results of a postmortem analysis, in which a massive accumulation of CD68+ cells was observed in the lungs of a patient with fatal M. tuberculosis–associated IRIS.36
Although the insights on the pathogenesis of immune reconstitution disease emerging from this study may have important implications for the understanding of mycobacterial-associated IRIS, the model we used does not fully explain all of the diverse manifestations of this disease, which can occur after ART treatment of HIV+ patients. For example, while our murine model uses active infection with M avium, the scenario of paradoxical IRIS is typically seen in patients whose IRIS-associated coinfections have been successfully treated. In addition, while IRIS occurs preferentially in patients beginning ART with low CD4 T-cell counts, these individuals, in contrast to our TCRα−/− mice, are not totally lymphopenic at the time of reconstitution, and thus it is possible that preexisting Ag-specific T cells may contribute to their disease induction. If so, disease induction by these Ag-experienced cells might be more dependent on lymphopenia-driven expansion than the disease triggered by naive T cells in the version of our model described here. Nevertheless, the possible influence of these parameters can be investigated in future studies with the model by using drug-treated M avium infection or by varying the number and activation status of the CD4 T cells used to mediate disease (ie, naive, memory, or tolerant CD4 T cells). Such studies may provide further fundamental insights into mechanisms that determine whether CD4 T cells become pathogenic or protective when reconstituting lymphopenic hosts as well as lead to hypotheses concerning the pathogenesis of IRIS that can be tested in clinical studies.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We are grateful to Tom Moyer and Carol Henry for performing the FACS cell purifications and to Dr Kevin Holmes and David Stephany for their advice on multicolor flow cytometry. We also thank Dr Antonio Rothfuchs for providing the stock of M avium and Drs Joel Ernst and Rachael Caspi for supplying mouse strains used in our experiments. Finally, we gratefully acknowledge Drs Nevil Singh, Carl Feng, Bruno Andrade, Beth Wohlfert, and Siamon Gordon for their invaluable advice and discussions.
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
National Institutes of Health
Authorship
Contribution: D.L.B. designed the research, performed the experiments, analyzed the data, made the figures, and wrote the manuscript; K.D.M.-B. designed the research and performed the experiments; L.R.V.A., M.S.W., S.W., P.C., and S.H. performed experiments; I.S. designed the research; and A.S. designed the research and wrote the manuscript.
The current affiliation for L.R.V.A. is Laboratory of Immunopathology, René Rachou Research Center, FIOCRUZ, Belo Horizonte, MG, Brazil. The current affiliation for M.S.W. is Division of Molecular Immunology, National Institutes for Medical Research, Mill Hill, London, United Kingdom.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daniel L. Barber, Immunology Section, Laboratory of Parasitic Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, 50 South Dr, Rm 6146, Bethesda, MD 20892; e-mail: barberd@niaid.nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal