

Abstract
Extracorporeal photochemotherapy (ECP) is widely used to treat cutaneous T-cell lymphoma, graft-versus-host disease, and allografted organ rejection. Its clinical and experimental efficacy in cancer immunotherapy and autoreactive disorders suggests a novel mechanism. This study reveals that ECP induces a high percentage of processed monocytes to enter the antigen-presenting dendritic cell (DC) differentiation pathway, within a single day, without added cytokines, as determined by enhanced expression of relevant genes. The resulting DCs are capable of processing and presentation of exogenous and endogenous antigen and are largely maturationally synchronized, as assessed by the level of expression of costimulatory surface molecules. Principal component analysis of the ECP-induced monocyte transcriptome reveals that activation or suppression of more than 1100 genes produces a reproducible distinctive molecular signature, common to ECP-processed monocytes from normal subjects, and those from patients. Because ECP induces normal monocytes to enter the DC differentiation pathway, this phenomenon is independent of disease state. The efficiency with which ECP stimulates new functional DCs supports the possibility that these cells participate prominently in the clinical successes of the treatment. Appropriately modified by future advances, ECP may potentially offer a general source of therapeutic DCs.
Introduction
Extracorporeal photochemotherapy (ECP) was originally designed as blood-directed palliative chemotherapy for lymphocytic leukemias, in which the natural plant product 8-methoxypsoralen (8-MOP) is ultraviolet-activated to a transient state that covalently crosslinks thymine bases of DNA.1 Surprisingly, in the original phase 1/2 clinical trial, in which less than 5% of the body's circulating malignant T cells were extracorporeally exposed to activated 8-MOP, 8-MOP stimulated clinically significant persistent immunologic responses to the reinfused damaged malignant T cells.2
Since that serendipitous finding, the world-wide accelerating use of ECP has generated clinical responses in cancer (cutaneous T-cell lymphoma [CTCL]), organ transplant recipients, and acute and chronic graft-versus-host disease (GVHD).3,4 These high clinical success rates, in otherwise therapeutically resistant situations, coupled with low toxicity and no recognized autoimmune sequelae,5 have led clinicians in tertiary care centers throughout the United States and Europe to administer ECP cell therapy one and a half million times. However, the fundamental principles underlying these clinical responses, involving immunostimulation in CTCL and immunosuppression and regulation in GVHD, have remained elusive, despite extensive scientific investigation in specifically designed experimental systems.4,6
A recent potentially elucidating insight into ECP's mechanism was the finding that the extracorporeal routing of patient blood through the 1-mm space between the ultraviolet exposure plates caused large scale conversion of passaged blood monocytes, from treated CTCL subjects, into leukocytes with features typical of antigen-presenting dendritic cells (DCs).7 This observation raised the possibility that ECP's therapeutic efficacy in CTCL may result from loading of newly formed DCs with antigenic apoptotic malignant T cells. In a pilot clinical trial, the improved clinical responses in CTCL patients resistant to conventional ECP, obtained by incorporating overnight coincubation of the new DCs with treated CTCL cells, prior to their reinfusion,8 empirically supported this premise. While augmenting cell-to-cell contact between injured CTCL cells and new DCs, that logical modification of the standard ECP procedure appeared to maintain the advantageous safety profile typical of conventional ECP.
Clarification of the mechanism of ECP is important. If ECP efficiently transforms processed blood monocytes into functional DCs, the procedure may mimic and amplify physiologic signaling through which this conversion normally occurs.9 If the clinical successes of ECP reflect production and antigen-loading of maturationally synchronized DCs, it may offer an especially safe and efficient method for additional immunotherapeutic purposes. If the resulting DCs can be extracorporeally influenced to preferentially stimulate either antigen-specific responses or suppression, it may become possible to tailor ECP to particular clinical situations.
This study demonstrates that passage of human monocytes through the extracorporeal exposure field stimulates expression of transcripts typical of DC differentiation as part of a highly distinctive transcriptome. The induced DCs are maturationally synchronized and effective antigen-presenting cells. ECP activation of a battery of genes encoding DC maturation supports the premise that the noted DC phenotype results from expedited monocyte-to-DC differentiation.
Methods
Patient samples
Leukocytes from patients undergoing ECP using the UVAR XTS Photopheresis System (Therakos) were obtained under the guidelines of the Yale Human Investigational Review Board. Informed consent was provided according to the Declaration of Helsinki. Aliquots were procured at 3 time points: before treatment (Pre ECP), immediately after 8-MOP/ultraviolet A (UVA) exposure (ECP Day 0) or after 18-hour incubation of treated blood mononuclear leukocytes (ECP Day 1) in a 1-L platelet storage bag (PL-2410; Baxter).
Normal subjects
To determine whether ECP induces monocytes from healthy subjects to convert to DCs, mononuclear leukocytes from normal subjects were examined in 2 ways. Leukapheresed leukocytes from normal subjects (N = 3) were studied pretreatment (pre-ECP), immediately after ECP (ECP Day 0), and 18 hours after ECP (ECP Day 1). A desktop apparatus, incorporating a UVA light source and a plastic exposure plate, enabled laboratory reproduction of the clinical ECP system and sample access for parallel RNA isolation, immunophenotyping, and functional studies. Alternatively, a unit of blood from normal subjects was drawn into a transfer bag and passed through the ECP treatment apparatus in an identical fashion to that of treated patients (N = 3). The cells obtained from the unit of normal blood were used for microarrays and antigen presentation assays.
Psoralen addition
As is routinely done during ECP, the standard 8-MOP concentrated solution (Therakos) was added directly to the clinical ECP apparatus and to the laboratory model system. That mode of introduction enabled precise 100-200 ng/mL concentrations throughout the clinical procedures and experimentation.
Overnight culture
In ECP, it is not possible to examine phenotypic and functional changes in treated monocytes, because those cells are immediately reinfused into patients. Therefore, after ECP, cells were cultured for 18 hours (RPMI 1640/15% autologous serum) to study induced monocyte gene activation, maturation and function. Prior to (pre-ECP) and immediately after ECP (ECP Day 0), patient and normal subject samples were isolated by centrifugation over a Ficoll-Hypaque gradient. The cells were resuspended in RPMI-1640 medium (Gibco), supplemented with 7.5% AB serum, 7.5% autologous serum (Gemini Bio-Products) and cultured (for patients) in 6-well polystyrene tissue-culture plates at a density of 5 × 106 cells/mL and in Baxter platelet storage bags (for normal subjects 37°C, 5% CO2). After overnight culture (ECP Day 1), cells were harvested before undergoing monocyte enrichment. To generate DCs for comparative phenotypic analysis, cells were cultured in RPMI 1640 15% serum in the presence of 1 mL of GMCSF and IL4 (25 ng/mL; R&D Systems) for 6 days.
Magnetic bead enrichment of the monocyte population
To enable determination of whether ECP activates genes directing monocytes into the DC maturational pathway, it was necessary to develop a gentle negative monocyte enrichment method that eliminates contribution of lymphocytes to the transcriptome analysis while minimizing monocyte physical or cell membrane perturbation. Monocytes were enriched from the mononuclear cell pool by single passage through affinity columns. This negative selection method limited physical perturbation, whereas lymphocytes adherent to magnetic microbeads (Miltenyi Biotec), conjugated to relevant monoclonal antibodies (anti-CD4, CD8, CD19), were depleted. However, enrichment of ECP Day 1 monocytes beyond 60%-80% proved challenging, because diminished surface display of lymphocyte markers by ECP-damaged lymphocytes permitted a fraction of T and B cells to escape retention in the columns. Repetitive passes through the affinity column, to further enhance monocyte purity, was not an option because that approach compounds the physical perturbation of passively filtered monocytes. Fortuitously, a series of analyses revealed that ECP's preferential damage of lymphocytes precluded the necessity of full purification of monocytes for accurate assessment of level of DC gene activation. Due to their extreme sensitivity to UVA-activated 8-MOP,10 99% of ECP-processed lymphocytes were apoptotic after overnight incubation (as determined by staining with APO2-PE, Trypan blue, and/or annexin–fluorescein isothiocynate FITC/propidium idodide). Because ECP causes global lymphocyte apoptosis, 90%-95% of viable mononuclear leukocytes in the ECP day 1 fraction were monocytes. This phenomenon accounts for the observation (discussed more fully in “Results”) that multiple step magnetic bead removal of apoptotic lymphocytes, performed as follows and yielding monocyte purity of greater than 95%, does not alter levels of observed gene expression in the studied cell populations. To accomplish that comparison we modified the monocyte purification procedure by adapting a negative selection protocol using magnetic beads and the EasySep magnet. Peripheral blood mononuclear cells were centrifuged at low speed (120g for 10 minutes) to remove platelets. Cells were then labeled using the Monocyte Isolation Kit II (Miltenyi Biotec) following the manufacturers procedure with the following modifications: (1) buffer consisted of ice-cold phosphate-buffered saline containing 2% autologous serum and 1mM EDTA (ethylenediaminetetraacetic acid); (2) blocking time was increased to 10 minutes; (3) labeling with the Biotin-Antibody Cocktail was increased to 20 minutes; and (4) cells were washed once between labeling with the Biotin-Antibody Cocktail and the Anti-Biotin Microbeads. To avoid stimulating the monocytes by passing them over a column, the magnetically labeled cells were instead separated from the unlabeled monocytes using the EasySep magnet (StemCell Technologies). Cells, in 2 mL of buffer in a 5-mL polystyrene tube, were placed in the magnet for 10 minutes, and then the unlabeled cells were carefully poured off into a new tube. This procedure was repeated 2×, to maximally enhance monocyte purity. At this point, because the purity was still insufficient, cells were relabeled with the Monocyte Isolation Kit II reagents and placed in the EasySep magnet for an additional 10 minutes, and the unlabeled monocytes were eluted. Final purity (X = 96%+4.5) was assessed by flow cytometric analysis of CD14 staining.
Immunophenotyping
Monoclonal antibodies specific for monocytes and DCs, included: CD14 (lipopolysaccharide [LPS] receptor, monocytes); CD36 (receptor for apoptotic cells, monocytes); human leukocyte antigen DR-1 (HLA-DR; class II major histocompatibility complex [MHC] molecule); CD83 (DC marker); cytoplasmic DC-lysosome–associated membrane protein (DC-LAMP; DC marker); and CD80 and CD86 (B7.1 and B7.2 costimulatory molecules).11-13 Antibodies were obtained from Beckman Coulter and used at their predetermined optimal dilutions. Background staining was established with appropriate isotype controls, and immunofluorescence was analyzed using a FC500 flow cytometer (Beckman Coulter). Combined membrane and cytoplasmic staining was performed following manufacturer's instructions for cell fixation and permeabilization (Intraprep kit; Beckman Coulter).
Antigen presentation assay
Volunteer freshly isolated, magnetic bead-enriched, antigen-experienced CD4+ populations (2 × 106/mL, 50 μL/well) were added to monocytes (2 × 106/mL, 50 μL/well) in the presence of tetanus toxoid (10 μg/mL, 100 μL/well) and RPMI medium 1640/15% autologous serum. After 5 days of culture, the cells received 1 μCi of [3H]-thymidine and were incubated overnight, harvested, and counted in a Beta liquid scintillation counter (PerkinElmer). Results are presented as the mean and standard deviation of 5 replicate cultures.
MLR/CML assay
To assess whether ECP-processed monocytes are functionally capable of stimulating MHC class I–restricted cytotoxicity by CD8 T cells, mononuclear leukocytes from 3 normal subjects were studied. One unit of anticoagulated blood, freshly procured from each of 3 HLA-A2–postive volunteers, served as sources of stimulator monocyte/DCs, before and after being processed through the clinical ECP apparatus in a manner identical to the actual ECP procedure. Mononuclear fractions were isolated from the blood immediately prior to ECP processing (pre-ECP) and immediately after ECP (ECP D0). After gamma irradiation (3000 rad, Cesium source) to ensure unidirectional T-cell stimulation, the Pre ECP fraction was serially diluted in RPMI 1640/15% autologous serum, and 100 μL containing from 25 000 to 250 cells was plated in round-bottom microtiter plate wells, in 5 replicates. The ECP D0 fraction was incubated for 18 hours in large well plates and harvested by scraping the wells to free adherent cells. The resuspended cells were then serially diluted and plated as above. An A-2–negative normal donor served as the source of responder CD4 and CD8 T cells, purified by positive selection on Miltenyi magnetic bead columns (average purity 98%). Responder T cells (50 000/well in 100 μL) were then added to the wells containing either Pre-ECP or ECP-D0 stimulators, and the plates were cultured for 7 days at 37°C in a CO2 incubator. For target cells, the A-2–positive T-B hybridoma lymphoblast line, 174 × CWM.T114 (provided by Peter Cresswell, Yale University), was labeled with 51Cr and added to the MLR cultures at 104 cells/well. After 4-hour incubation, plates were centrifuged, and 100 μL of supernatant was removed from each well for counting in a gamma counter. “Percent-specific lysis” was defined as 100 times the following fraction:
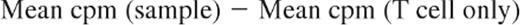
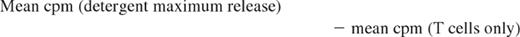
RNA isolation and microarray hybridization
Total RNA was isolated using RNeasy Mini Kit columns with on-column DNase I treatment (QIAGEN). RNA yield and purity were measured using the NanoDrop ND-1000 Spectrophotometer and the Agilent 2100 Bioanalyzer. Fragmented cRNAs were hybridized on Affymetrix HG U133 Plus 2.0 human chips, and screening for approximately 47 400 human genes and ESTs was performed by the Yale University W. M. Keck Resource Laboratory. The microarray results are available on Gene Expression Omnibus under accession number GSE23604.
Data analysis
Raw data without normalization generated from Affymetrix GeneChip Operating Soft-ware Version 1.2 (GCOS 1.2; Affymetrix) were analyzed using GeneSpring software 7.2 (Agilent Technologies-Silicon Genetics). Data were normalized using Robust Multi-Array. Only probe sets with a minimal fold change of > 2.0 combined with an average signal intensity of 500 or higher in either leukapheresis or treated samples were included in the analysis. Differential gene expression was considered as a ≥ 2-fold change and P ≤ .05. Principal component analysis (PCA) of the induced transcriptomes was performed by standard methodology.15 Signal transduction pathway involvement was identified with MetaCore Software Version 1.0 (GeneGo).
Quantitative real-time PCR
Microarray expression of selected genes was confirmed in aliquots of the same RNA samples, using quantitative real-time polymerase chain reaction (PCR). RNA was reverse transcribed to cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Reverse transcription was carried out in a 96-well thermocycler (MJ Research PTC-200) in the following conditions: 25°C, 10 minutes, 37°C, 120 minutes, 85°C, 5 seconds. TaqMan real-time PCR was used to detect transcripts of DC-LAMP, CCR7, CD80, CD86, and CD14. Primers and probes for each sequence were obtained as inventoried Taqman Gene Expression Assays (Applied Biosystems). HPRT1 was used as a reference gene.
Results
Phenotypic evidence for a monocyte to DC transition
We first determined whether our previous preliminary finding, that ECP induces a dendritic cell phenotype on ECP-processed monocytes from CTCL patients,7 is a general phenomenon or merely limited to the CTCL clinical setting. We assessed coexpression of HLA-DR (abundantly displayed on DCs) with induced cytoplasmic CD83, a marker of early DC maturation used in that prior study, in monocytes procured from each of 3 CTCL patients, 3 GVHD patients and 3 normal subjects. The patient monocytes were obtained from the initial leukapheresis phase of ECP (pre ECP), and then processed through the actual online ECP exposure plate (ECP Day 0). The normal subject monocytes were obtained from an equivalent leukapheresis of normal subjects and were then processed through the laboratory equivalent of the ECP exposure plate.
As shown in Figure 1A, in all instances large increases in the percentage of processed monocytes, coexpressing HLA-DR and cytoplasmic CD83, were identified after 18-hour incubation of the treated cells (ECP Day 1). Overnight incubation produced 34% monocyte-to-DC conversion, an 8-fold increase in the expression of this standard DC marker over background levels.

ECP generation of dendritic cells. The percentage and absolute number of DCs generated in ECP-processed monocytes. (A) The percentage of monocytes induced by ECP to enter the DC differentiation pathway was assessed by coexpression of cytoplasmic CD83 and cell-surface HLA-DR. Two-color flow cytometric analysis of 10 000 monocytes from 3 CTCL and 3 GVHD patients and from 3 normal subjects is shown at the 3 time points: from the leukapheresis harvest prior to ECP (pre ECP), immediately after ECP (ECP Day 0), and 18 hours after ECP (ECP Day 1). The monocyte population (confirmed by CD11c staining) was gated using forward and side scatter. Coexpression of cytoplasmic CD83 and cell membrane HLA-DR was assessed in the gated monocyte population. The last set of bars demonstrates the mean ± SD for all 9 subjects, revealing significant enhancement in CD83 expression after 18-hour incubation (P < .001), compared with pre- and immediately post-ECP. (B) The absolute number of ECP-processed monocytes that entered the DC differentiation pathway was determined for each test subject. The absolute number of DCs was calculated as the product of 2 numbers: (1) the percentage of leukocytes cytometrically gating, by forward and side scatter, in the Monocyte/DC region and (2) the total number of cells in that region displaying the CD11c marker typical of both monocytes and DCs. The absolute number of induced CD83+ cells varied with white blood cell count of the subject, but exceeded 50 million in all but 1 CTCL patient and exceeded 300 million in 1 normal subject. The absolute number of DCs was then calculated by multiplying the CD83+ percentage by the relevant total volume.
ECP generation of dendritic cells. The percentage and absolute number of DCs generated in ECP-processed monocytes. (A) The percentage of monocytes induced by ECP to enter the DC differentiation pathway was assessed by coexpression of cytoplasmic CD83 and cell-surface HLA-DR. Two-color flow cytometric analysis of 10 000 monocytes from 3 CTCL and 3 GVHD patients and from 3 normal subjects is shown at the 3 time points: from the leukapheresis harvest prior to ECP (pre ECP), immediately after ECP (ECP Day 0), and 18 hours after ECP (ECP Day 1). The monocyte population (confirmed by CD11c staining) was gated using forward and side scatter. Coexpression of cytoplasmic CD83 and cell membrane HLA-DR was assessed in the gated monocyte population. The last set of bars demonstrates the mean ± SD for all 9 subjects, revealing significant enhancement in CD83 expression after 18-hour incubation (P < .001), compared with pre- and immediately post-ECP. (B) The absolute number of ECP-processed monocytes that entered the DC differentiation pathway was determined for each test subject. The absolute number of DCs was calculated as the product of 2 numbers: (1) the percentage of leukocytes cytometrically gating, by forward and side scatter, in the Monocyte/DC region and (2) the total number of cells in that region displaying the CD11c marker typical of both monocytes and DCs. The absolute number of induced CD83+ cells varied with white blood cell count of the subject, but exceeded 50 million in all but 1 CTCL patient and exceeded 300 million in 1 normal subject. The absolute number of DCs was then calculated by multiplying the CD83+ percentage by the relevant total volume.
This data revealed that the development of a DC phenotype is not limited to ECP-treated monocytes from CTCL patients, but also applies to those from GVHD patients and normal subjects. The absolute number of ECP-processed monocytes that expressed this DC phenotype after overnight incubation was assessed in cells from 6 patients and 3 normal subjects (Figure 1B). Electron microscopic examination confirmed the production of morphologically typical viable DCs able to bind, internalize, and digest apoptotic lymphocytes.
Using coexpression of HLA-DR and CD83 positivity as the identity criterion, the 1-day DC yield from ECP processing of monocytes from normal subjects (54-316 × 106) compares favorably to the total DC harvest from 6-day conventional GM-CSF/IL4 maturation of monocytes procured from full leukapheresis.16
Induced conversion of monocytes-to-DCs in normal, CTCL, and GVHD subjects
Representative flow cytograms (Figure 2) demonstrate ECP-induced monocyte-to-DC conversion for monocytes procured from CTCL and GVHD patients. Importantly, this phenomenon is a general one, not limited to those disease settings, because ECP also induced monocytes from normal subjects to express the same DC markers. After overnight incubation of the ECP cells, the monocyte/DC populations from each of these sources increased membrane expression of the costimulatory molecules CD80 and CD86 (B7.1 and 2), and the DC cytoplasmic markers CD83 and DC-LAMP (significant in CTCL patients and normal subjects).

ECP-induction of costimulatory molecules DC-LAMP and class II MHC was determined by flow cytometric analysis of processed monocytes. Monocytes were from (A) CTCL, (B) normal subjects, and (C) GVHD patients. Forward and side scatter gating, confirmed by CD11c and CD14 staining, was used to identify the monocyte population. The cells were fixed and permeabilized and the monocyte/DC population was stained for HLA-DR (FITC) on the membrane and CD83 or DC-LAMP (phycoerythrin, PE) in the cytoplasm to identify differentiating DCs. Two-color membrane staining for CD80 (FITC) and CD86 (PE) was used to identify expression of costimulatory molecules. Results are presented from 1 representative subject per group.
ECP-induction of costimulatory molecules DC-LAMP and class II MHC was determined by flow cytometric analysis of processed monocytes. Monocytes were from (A) CTCL, (B) normal subjects, and (C) GVHD patients. Forward and side scatter gating, confirmed by CD11c and CD14 staining, was used to identify the monocyte population. The cells were fixed and permeabilized and the monocyte/DC population was stained for HLA-DR (FITC) on the membrane and CD83 or DC-LAMP (phycoerythrin, PE) in the cytoplasm to identify differentiating DCs. Two-color membrane staining for CD80 (FITC) and CD86 (PE) was used to identify expression of costimulatory molecules. Results are presented from 1 representative subject per group.
Homogeneity of the ECP-induced DC population is indicated by the relatively uniform increase in CD86 (B7.2) expression (Figure 3A), compared with the more heterogeneous expression of costimulatory molecule on DCs generated by 6 days incubation with GMCSF/IL4 (Figure 3B). Together, these results extend our previous studies in CTCL,7 by demonstrating that monocytes from normal subjects and patients enter the DC differentiation pathway after ECP.

The rapidity of ECP induction of costimulatory molecule CD86 by monocytes, along with the homogeneity of its expression, was compared in parallel with the efficiency of the conventional method. (A) This typical flow cytometric histogram of a normal subject's ECP-processed monocyte population reveals the rapidity with which homogeneous expression of the costimulatory molecule CD86 (B7.2) was induced. After 18-hour incubation (Day 1), 94% of ECP-processed monocytes uniformly increased their expression of the cell surface CD86 (red), above the level of the isotype control (blue). Gating of leukocytes into the Monocyte/DC fraction was accomplished by forward and side scatter. Mean fluorescence intensity (MFI) of the induced CD86+ cells was 15. With such a large fraction of processed monocytes positive, the overall MFI was nearly as high (14.5). (B) ECP induction of CD86 was more rapid and extensive than that observed with cytokine-stimulated conventional monocyte-to-DC conversion. Even after the usual 6 days of culture with GMCSF and IL4, cytometric analysis of monocytes from a typical normal subject revealed less uniform induction of this costimulatory molecule. Because, for immunotherapeutic protocols, antigen loading of DC produced in this manner occurs at this juncture, prior to a second maturation step, this time point is most appropriate for comparison with ECP-induced DCs. The percentage of monocytes expressing CD86 (64 %) was lower than that observed with ECP-processed monocytes after only a single day of incubation. Although the MFI of the positive fraction was moderately higher at 6 days (MFI = 20) than that of the ECP-induced DCs at 1 day, the overall MFI was lower (MFI = 10.8), with nearly one-third of monocytes remaining negative for this marker.
The rapidity of ECP induction of costimulatory molecule CD86 by monocytes, along with the homogeneity of its expression, was compared in parallel with the efficiency of the conventional method. (A) This typical flow cytometric histogram of a normal subject's ECP-processed monocyte population reveals the rapidity with which homogeneous expression of the costimulatory molecule CD86 (B7.2) was induced. After 18-hour incubation (Day 1), 94% of ECP-processed monocytes uniformly increased their expression of the cell surface CD86 (red), above the level of the isotype control (blue). Gating of leukocytes into the Monocyte/DC fraction was accomplished by forward and side scatter. Mean fluorescence intensity (MFI) of the induced CD86+ cells was 15. With such a large fraction of processed monocytes positive, the overall MFI was nearly as high (14.5). (B) ECP induction of CD86 was more rapid and extensive than that observed with cytokine-stimulated conventional monocyte-to-DC conversion. Even after the usual 6 days of culture with GMCSF and IL4, cytometric analysis of monocytes from a typical normal subject revealed less uniform induction of this costimulatory molecule. Because, for immunotherapeutic protocols, antigen loading of DC produced in this manner occurs at this juncture, prior to a second maturation step, this time point is most appropriate for comparison with ECP-induced DCs. The percentage of monocytes expressing CD86 (64 %) was lower than that observed with ECP-processed monocytes after only a single day of incubation. Although the MFI of the positive fraction was moderately higher at 6 days (MFI = 20) than that of the ECP-induced DCs at 1 day, the overall MFI was lower (MFI = 10.8), with nearly one-third of monocytes remaining negative for this marker.
Functional competence of ECP-generated DCs
We next inquired whether ECP-processed antigen-presenting cells (APCs) are capable of stimulating normal T-cell responses to class I and II MHC-presented antigens. We first tested the capacity of ECP-induced DCs to present tetanus toxoid antigen to magnetic bead-enriched (but otherwise untreated) CD4+ T cells from tetanus-immune normal volunteers (Figure 4). Such T enriched cells, relatively depleted of antigen processing/presenting cells, exhibited only modest proliferative responses after 5 days of culture with tetanus toxoid. Furthermore, because T cells are lethally injured by exposure to ultraviolet-activated 8-MOP, ECP treated cells cultured in the presence of tetanus antigen produced a negligible proliferative response. However, the strikingly enhanced response to tetanus observed in cocultures containing both enriched CD4+ T cells and ECP-treated cells clearly demonstrates that the ECP-treated population functions effectively as a source of antigen processing and presenting DCs. These results show that the ECP-induced DCs, unlike the apoptotic coprocessed T cells, remain viable and capable of stimulating CD4 T-cell responses.

ECP-induced DCs efficiently processed and presented tetanus toxoid antigen to responsive autologous CD4 T cells. ECP-induced DCs, from a tetanus-immunized normal subject, efficiently processed and presented tetanus toxoid antigen to fresh autologous magnetic bead-purified CD4 T cells. Whereas autologous CD4 T cells alone or ECP-treated mononuclear cells (containing 8-MOP totally inhibited T cells) yielded limited or no response, respectively, addition of purified fresh CD4 T cells (third bar) enabled a brisk antigen-specific response. This result is representative of the effective presentation of tetanus antigen by ECP-induced DCs from each of 3 normal subjects.
ECP-induced DCs efficiently processed and presented tetanus toxoid antigen to responsive autologous CD4 T cells. ECP-induced DCs, from a tetanus-immunized normal subject, efficiently processed and presented tetanus toxoid antigen to fresh autologous magnetic bead-purified CD4 T cells. Whereas autologous CD4 T cells alone or ECP-treated mononuclear cells (containing 8-MOP totally inhibited T cells) yielded limited or no response, respectively, addition of purified fresh CD4 T cells (third bar) enabled a brisk antigen-specific response. This result is representative of the effective presentation of tetanus antigen by ECP-induced DCs from each of 3 normal subjects.
We next determined whether ECP-generated APCs, procured after processing blood from 3 normal volunteers through the clinical ECP apparatus, were also capable of mediating cytotoxicity by targeting HLA on appropriate lymphoblasts. To ensure that the results directly reflect functional properties of those APCs generated by ECP and intravenously returned to treated patients, we procured a unit of anti-coagulated blood from each normal volunteer and processed that blood through the actual ECP apparatus in a manner exactly equivalent to that of the clinical procedure. The experimental design sequentially involved ECP generation of APCs from HLA-A2 donors, use of those A2-positive APCs to stimulate in vitro allogeneic normal CD4 and CD8 responder cells and determination of the capacity of the allo-stimulated T cells to then lyse A2-positive lymphoblasts. The ability of the ECP-generated APC to induce these cytotoxic T-cell responses was tested over serial dilutions (ranging from 1:2 to 1:200 stimulator-to-responder ratios). Significant cytotoxicity was observed at each of the 3 highest ratios of ECP-generated APCs to responder cells. A representative titration of this phenomenon is shown in Figure 5, where it is noteworthy that ECP-APCs more effectively stimulated cytotoxic T-cell responses than did pretreatment APCs from the same normal donor at similar stimulator-to-responder ratios.

ECP induced APC efficiently stimulate T-cell mediated anti-class I MHC cytotoxicity. Normal donor whole blood was processed through the clinical ECP apparatus to determine whether the procedure yields APCs capable of stimulating vigorous CD8-mediated cytotoxicity. This representative example, from 3 parallel experiments involving different normal donors, shows that APCs from an HLA-A2–positive donor initiated allogeneic normal HLA-A2–negative T cells to potently target HLA-A2-positive lymphoblasts. Cytotoxic T-cell responses were tested over a broad range of stimulator-to-responder ratios. Most prominently at ratios of 1:6 and 1:20, the ECP-generated APCs more effectively stimulated cytotoxic T-cell responses than did autologous pretreatment monocytes. These results indicate that the ECP-generated APCs are functionally capable of initiating CD8 T-cell targeting of class I MHC.
ECP induced APC efficiently stimulate T-cell mediated anti-class I MHC cytotoxicity. Normal donor whole blood was processed through the clinical ECP apparatus to determine whether the procedure yields APCs capable of stimulating vigorous CD8-mediated cytotoxicity. This representative example, from 3 parallel experiments involving different normal donors, shows that APCs from an HLA-A2–positive donor initiated allogeneic normal HLA-A2–negative T cells to potently target HLA-A2-positive lymphoblasts. Cytotoxic T-cell responses were tested over a broad range of stimulator-to-responder ratios. Most prominently at ratios of 1:6 and 1:20, the ECP-generated APCs more effectively stimulated cytotoxic T-cell responses than did autologous pretreatment monocytes. These results indicate that the ECP-generated APCs are functionally capable of initiating CD8 T-cell targeting of class I MHC.
Collectively, these findings reveal that ECP rapidly induces a large fraction of monocytes to assume phenotypic properties typical of DCs. The ECP-generated APCs are viable and capable of efficiently processing and presenting recall antigens to responsive CD4 T cells, and stimulating cytotoxic T cells against class I MHC targets.
ECP induction of a distinctive transcriptome in processed monocytes
To determine whether ECP-induction of DCs reflects organized direction of a high fraction of monocytes into the DC differentiation pathway, we studied differential gene expression in cells from 6 normal subjects, and from 3 ECP-treated CTCL and 3 GVHD patients. ECP-induced alteration of gene expression was quantified as the fold change in RNA transcript levels from the pre-ECP baseline. In ECP, the processed monocytes are sequentially influenced by ex vivo factors: centrifugal force (during the leukapheresis procurement phase), interaction with the flat plastic surface and shearing forces (while being pumped through the UVA exposure plate as a 1-mm film), and exposure in that plate to the transiently photoactivated DNA-cross-linking 8-MOP. To test the premise that ECP induces functional DCs, we searched for a distinctive molecular signature in the more than 2500 genes either activated or suppressed by the 3-hour procedure.
Principal component analysis (PCA)15 is a well-established mathematical method that enabled us, through use of approximately 40 000 genome probes, to reduce the transcriptomes of each processed monocyte population to a single graphically represented point, for broad comparison with the transcriptomes of ECP-processed monocytes from other normal subjects or patients. As shown in Figure 6, ECP induced remarkably consistent clusters of affected genes (shown as ellipses accounting for the relevant sample group with a P < .05), regardless of whether the analyzed monocytes were obtained from normal subjects or patients (CTCL or GVHD). After overnight incubation, the transcriptomes of ECP processed monocytes, from both normal and diseased individuals, clustered tightly and distinctively together. This finding reveals that ECP induces a highly characteristic and nonrandom constellation of gene expression, worthy of the more focused individual gene analysis described below.

ECP reproducibly produced distinctive pan-genomic transcriptome activation in processed monocytes. Principal component analysis (PCA) of global gene expression data are graphed, using the 2 principal variance components. Each symbol represents the entire transcriptome for a single sample (N = 34: 11 pre ECP (red), 11 ECP Day 0 (green), and 11 from ECP-processed monocytes Day 1 (blue). The position of each point is calculated as compression of the full set of 40 000 data points, generated using the pan-genomic battery of probes: cells obtained from normal subjects (triangles), CTCL patients (circles), and GVHD patients (squares). The analysis reveals that the samples segregate based on treatment irrespective of whether the processed monocytes derived from patients or normal subjects. The data grouping is highlighted by the ellipses in lighter red for the pre ECP, green for the immediately post-ECP, and blue for the post-18–hour ECP monocytes. This data reveal that ECP's complex impact on the monocyte transcriptome is high predictable and distinctive after 18-hour incubation.
ECP reproducibly produced distinctive pan-genomic transcriptome activation in processed monocytes. Principal component analysis (PCA) of global gene expression data are graphed, using the 2 principal variance components. Each symbol represents the entire transcriptome for a single sample (N = 34: 11 pre ECP (red), 11 ECP Day 0 (green), and 11 from ECP-processed monocytes Day 1 (blue). The position of each point is calculated as compression of the full set of 40 000 data points, generated using the pan-genomic battery of probes: cells obtained from normal subjects (triangles), CTCL patients (circles), and GVHD patients (squares). The analysis reveals that the samples segregate based on treatment irrespective of whether the processed monocytes derived from patients or normal subjects. The data grouping is highlighted by the ellipses in lighter red for the pre ECP, green for the immediately post-ECP, and blue for the post-18–hour ECP monocytes. This data reveal that ECP's complex impact on the monocyte transcriptome is high predictable and distinctive after 18-hour incubation.
Large ECP-induced changes in individual gene expressions
The stimulation by ECP of individual gene activation in monocytes was expressed as the ratio of ECP Day 1 to pre-ECP expression for the relevant gene. To preclude inadvertent gene induction during monocyte enrichment, a negative column purification method was used, whereby lymphocytes were retained, and monocytes were passively filtered. The results revealed that the ECP-processed monocytes from both patients and normal subjects remain sufficiently viable to reproducibly express a shared transcriptome signature.
Genes were considered significantly up- or down-regulated by ECP if fold change was > 2 and significance was P < .05 compared with pre ECP. Levels of RNA transcripts from approximately 3000 genes were significantly changed in each patient group and in normal subjects (Table 1). Overall, 1129 genes were up- or down-regulated in common by ECP-processed monocytes from both CTCL and GVHD patients and from normal subjects, indicating commonality in ECP-induced gene activation.
Number of genes with altered expression after ECP
| Monocyte source | Total | Up-regulated | Down-regulated |
|---|---|---|---|
| Normal subjects (alone): N = 6 | 2820 | 1111 (39%) | 1709 (61%) |
| CTCL (alone): N = 3 | 3178 | 1956 (62%) | 1222 (38%) |
| GVHD (alone): N = 3 | 3235 | 2008 (62%) | 1227 (38%) |
| CTCL/GVHD (shared): N = 6 | 2532 | 1474 (58%) | 1058 (42%) |
| Normal/CTCL/GVHD (shared): N = 12 | 1129 | 498 (44%) | 631 (56%) |
| Monocyte source | Total | Up-regulated | Down-regulated |
|---|---|---|---|
| Normal subjects (alone): N = 6 | 2820 | 1111 (39%) | 1709 (61%) |
| CTCL (alone): N = 3 | 3178 | 1956 (62%) | 1222 (38%) |
| GVHD (alone): N = 3 | 3235 | 2008 (62%) | 1227 (38%) |
| CTCL/GVHD (shared): N = 6 | 2532 | 1474 (58%) | 1058 (42%) |
| Normal/CTCL/GVHD (shared): N = 12 | 1129 | 498 (44%) | 631 (56%) |
Number of genes significantly induced or suppressed by ECP. Activity of 1129 genes was similarly affected by ECP, in mononuclear cells procured from normal subjects and patients (CTCL and GVHD).
Increased expression of numerous genes associated with DC differentiation, adhesion, and function (Tables 2–3) further support ECP stimulation of entry of monocytes into that pathway. DC-LAMP (lysosome-associated membrane glycoprotein),17 expressed during DC activation and differentiation, is thought to play a role in class II antigen processing and serves as a specific mature DC marker. Glycoprotein NMB is a glycoprotein that is up-regulated in mature DCs and involved in DC adhesion through an RGD site that binds integrins.18 CD80 and CD86 (costimulatory molecules, B7.1 and 7.2) ligate CD28 on T cells and, in concert with T-cell receptor triggering, activate antigen-responsive T cells.19
ECP-enhanced expression of DC marker genes ratio* of ECP D1/pre-ECP levels
| Gene | Attributes | CTCL and GVHD (N = 6) induced expression ratio | Normal subjects (N = 6) induced expression ratio |
|---|---|---|---|
| DC-LAMP | DC lysomal protein, role in class II antigen processing.17 | 27.6 | 17.2 |
| P = 1.2 × 10−9 | P = 1.4 × 10−7 | ||
| GPNMB | Transmembrane glycoprotein containing the integrin-binding motif RGD.18 | 205.7 | 123.3 |
| P = 9.6 × 10−15 | P = 2.8 × 10−14 | ||
| CD80 | Costimulatory molecule, B7.1 activates T cells.19 | 13.4 | NC |
| P = 2.3 × 10−13 | |||
| CD86 | Costimulatory molecule, B7.2 activates T cells.19 | NC | 50P = 1.4×10−5 |
| CD40 | Prolongs B7.1 and 2 T-cell activation.19 | 2.3 | NC |
| P = 5.7 × 10−4 | |||
| Decysin | Expressed in LPS-matured DCs but not monocytes or immature DCs.20 | 26.5 | 7.1 |
| P = 1.0 × 10−9 | P = 5.6 × 10−4 | ||
| CCR7 | Facilitates DC entry into lymph nodes where antigen presentation to naïve T cells occurs.21 | 2.6P = 7.0 × 10−3 | NC |
| CD83 | DC maturation molecule.11-13 | NC | 2.3 |
| P = .03 | |||
| OLR1 | Lox1, lectin-like receptor expressed on DCs but absent from monocytes.25 | 13.6 | 100.1 |
| P = 3.3 × 10−5 | P = 8.3 × 10−8 | ||
| FPRL2 | Formyl peptide receptor-like-2 expressed on mature DCs and involved in trafficking | 33.9 | 43.2 |
| and T-cell stimulation.26 | P = 2.1 × 10−8 | P = 1.9 × 10−8 |
| Gene | Attributes | CTCL and GVHD (N = 6) induced expression ratio | Normal subjects (N = 6) induced expression ratio |
|---|---|---|---|
| DC-LAMP | DC lysomal protein, role in class II antigen processing.17 | 27.6 | 17.2 |
| P = 1.2 × 10−9 | P = 1.4 × 10−7 | ||
| GPNMB | Transmembrane glycoprotein containing the integrin-binding motif RGD.18 | 205.7 | 123.3 |
| P = 9.6 × 10−15 | P = 2.8 × 10−14 | ||
| CD80 | Costimulatory molecule, B7.1 activates T cells.19 | 13.4 | NC |
| P = 2.3 × 10−13 | |||
| CD86 | Costimulatory molecule, B7.2 activates T cells.19 | NC | 50P = 1.4×10−5 |
| CD40 | Prolongs B7.1 and 2 T-cell activation.19 | 2.3 | NC |
| P = 5.7 × 10−4 | |||
| Decysin | Expressed in LPS-matured DCs but not monocytes or immature DCs.20 | 26.5 | 7.1 |
| P = 1.0 × 10−9 | P = 5.6 × 10−4 | ||
| CCR7 | Facilitates DC entry into lymph nodes where antigen presentation to naïve T cells occurs.21 | 2.6P = 7.0 × 10−3 | NC |
| CD83 | DC maturation molecule.11-13 | NC | 2.3 |
| P = .03 | |||
| OLR1 | Lox1, lectin-like receptor expressed on DCs but absent from monocytes.25 | 13.6 | 100.1 |
| P = 3.3 × 10−5 | P = 8.3 × 10−8 | ||
| FPRL2 | Formyl peptide receptor-like-2 expressed on mature DCs and involved in trafficking | 33.9 | 43.2 |
| and T-cell stimulation.26 | P = 2.1 × 10−8 | P = 1.9 × 10−8 |
Fold increase in expression of multiple genes involved in DC maturation and function induced by ECP.
Impact of treatment on gene expression is displayed as an induced expression ratio (ratio of ECP Day 1 to pre-ECP expression for the relevant gene). RNA was isolated from 3 CTCL patients and 3 GVHD patients and 6 normal subjects at the relevant time points.
ECP-enhanced expression of cell adhesion–associated genes ratio* of ECP Day 1/Pre-ECP levels
| Gene | Attributes normal | CTCL and GVHD (N = 6) induced expression ratio | Normal subjects (N = 6) induced expression ratio |
|---|---|---|---|
| Integrin β8 (ITGB8) | Expressed on DCs and plays a role in TGF-β activation to prevent immune dysfunction.27 | 26.3 | 4.2 |
| P = 5.0 × 10−12 | P = 1.2 × 10−5 | ||
| Integrin α5 (ITGA5) | Component of fibronectin receptor.28 | 3.1 | 3.1 |
| P = 1.3 × 10−5 | P = 7.4 × 10−5 | ||
| Integrin αV (ITGAV) | Component of vitronectin receptor.29 | 3.7 | 4.0 |
| P = 6.6 × 10−6 | P = 9.4 × 10−6 | ||
| Osteopontin (SPP1) | Regulates cytokine production in DCs.30 | 45.6 | 15.7 |
| P = 4.1 × 10−8 | P = 1.1 × 10−5 | ||
| ICAM 1 | Mediates DC interaction with T cells.31 | 3.2 | 3.7 |
| P = 1.3 × 10−5 | P = 7.2 × 10−4 | ||
| Matrix metalloprotease | |||
| MMP9 | Mature DC marker; contributes to migration.32 | 17.3 | 6.8 |
| P = 3.3 × 10−7 | P = 1.9 × 10−4 |
| Gene | Attributes normal | CTCL and GVHD (N = 6) induced expression ratio | Normal subjects (N = 6) induced expression ratio |
|---|---|---|---|
| Integrin β8 (ITGB8) | Expressed on DCs and plays a role in TGF-β activation to prevent immune dysfunction.27 | 26.3 | 4.2 |
| P = 5.0 × 10−12 | P = 1.2 × 10−5 | ||
| Integrin α5 (ITGA5) | Component of fibronectin receptor.28 | 3.1 | 3.1 |
| P = 1.3 × 10−5 | P = 7.4 × 10−5 | ||
| Integrin αV (ITGAV) | Component of vitronectin receptor.29 | 3.7 | 4.0 |
| P = 6.6 × 10−6 | P = 9.4 × 10−6 | ||
| Osteopontin (SPP1) | Regulates cytokine production in DCs.30 | 45.6 | 15.7 |
| P = 4.1 × 10−8 | P = 1.1 × 10−5 | ||
| ICAM 1 | Mediates DC interaction with T cells.31 | 3.2 | 3.7 |
| P = 1.3 × 10−5 | P = 7.2 × 10−4 | ||
| Matrix metalloprotease | |||
| MMP9 | Mature DC marker; contributes to migration.32 | 17.3 | 6.8 |
| P = 3.3 × 10−7 | P = 1.9 × 10−4 |
ECP-induced fold increase in expression of genes affecting monocyte adhesion, migration, and signaling.
Impact of treatment on gene expression is displayed as an induced expression ratio (ratio of ECP Day 1 to pre-ECP expression for the relevant gene). RNA was isolated from 3 CTCL patients and 3 GVHD patients and 6 normal subjects at the relevant time points.
CD80 gene expression was significantly up-regulated only in patients and CD86 only in normal subjects. However, display of both molecules was enhanced in all subjects by immunophenotyping (see Figure 2 for representative examples). This finding suggests that ECP stimulates transport of preformed costimulatory molecules to the DC surface, in addition to variable activation of the relevant genes.
As would be expected during monocyte-to-DC maturation, CD14 (monocyte marker) expression was diminished, as assessed by measuring the mean fluorescence intensity on the monocyte populations of all patients and normal subjects, after overnight culture of ECP-processed monocytes. This result was confirmed in RT-PCR studies of the patients' post-ECP cells (results not shown).
CD40, a costimulatory molecule expressed on mature DCs, interacts with CD40 ligand on activated T cells and may enhance or prolong B7-1 and 2 T-cell activation.19 Decysin, a disintegrin/metalloprotease, is increased in LPS-matured DCs and absent from monocytes and immature DCs,20 thereby serving as a marker of DC differentiation. CCR7 is a chemokine receptor required for DC migration to lymphoid organs, where the DCs present antigen to naive T cells.21
Increased mRNA expression of CD83, a marker of maturing DCs,11-13 was identified only in normal subjects, whereas enhanced CD83 protein expression was detected by immunophenotyping in the cytoplasm of differentiating DCs in all 3 groups (Figure 2). Possible explanations for increased cytoplasmic CD83 protein expression, in the absence of a uniform increase in CD83 mRNA expression in all patients and normal subjects, include (1) different kinetics, that is, an increase in mRNA expression may have occurred prior to the time RNA was harvested from the ECP Day 1 cells (after 18-hour culture); and (2) posttranscriptional regulation of CD83 mRNA, that is, the export of transcribed CD83 mRNA from the nucleus to its site of translation in the cytoplasm. This latter phenomenon has recently been shown to distinguish CD83 production and is tightly regulated (both positively and negatively) by several distinct enzymes and cofactors involved in nucleocytoplasmic shuttling.22-24 The posttranscriptional regulation could temporarily disconnect the expression of CD83 mRNA and production of CD83 protein.
Collectively, this transcriptional profiling reveals that cells from patients, as well as from normal subjects, activate or suppress multiple genes associated with differentiation of monocytes into maturing DCs.
RT-PCR demonstration of ECP activation of DC-distinctive genes in monocytes
Because of the necessity of avoiding monocyte activation in the process of enriching the studied mononuclear populations, we used a negative affinity column enrichment method in which lymphocytes were retained by adherence to anti–T- and B-cell monoclonal antibodies conjugated to magnetic beads. The level of monocyte enrichment obtainable by this approach was limited to 60%-80%, by factors and considerations intrinsic to our experimental design. ECP, through the impact of 8-MOP DNA cross-links, initiates virtually universal apoptosis in the processed highly sensitive lymphocytes.10 The resulting diminished surface expression of T- and B-cell differentiation antigens diminishes the affinity of these cells for affinity column, thereby limiting lymphocyte retention, leading to lymphocyte contamination of the passively transiting monocyte pool. Although our preliminary findings had suggested that gene activation in the apoptotic contaminating lymphocytes does not discernibly contribute to the finding of prominent induction of DC-distinctive genes, this possibility needed to be rigorously excluded.
For this purpose, we adapted the EasySep leukocyte enrichment method to enrich monocytes to > 95% in the tested populations, also utilizing monoclonal antibody–coated magnetic beads. Because this approach did not involve column passage, we found that, despite the necessity of multiple purification cycles to remove the very large majority of the ECP-damaged lymphocytes, inadvertent activation of the battery of distinctive DC genes is avoided. The availability of these more highly purified populations of ECP-processed monocytes then enabled us to confirm, by qRT-PCR, that the therapeutic procedure impressively stimulates expression of DC-distinctive genes.
Figure 7 demonstrates that expression of each of the 10 examined DC-distinctive genes was significantly enhanced by ECP processing. Eighteen hours after passage through the ECP device, highly purified monocytes augmented their activation of DC-distinctive genes between 3 and > 1000-fold over identically enriched pretreatment monocytes. The tight correlation between the gene activation ratios of purified monocyte and nonpurified mononuclear populations indicates that ECP-damaged lymphocytes do not discernibly contribute to the enhanced expression of these genes.

RT-PCR analysis confirmed that ECP broadly activates genes distinctive of monocyte maturation to DCs. Study of negatively enriched ECP-processed monocytes from 4 normal donors, to 95% population purity, confirmed the microarray finding that ECP rapidly initiates monocyte entry into the DC pathway. Expression of each of 11 examined DC-distinctive genes was significantly enhanced by ECP processing, 3 and > 1000-fold over that manifested by pretreatment monocytes. Nearly indistinguishable findings from identical analysis of nonpurified mononuclear populations indicates that ECP-lethally damaged lymphocytes do not discernibly contribute to the observed expression of these genes, even when present in the study population.
RT-PCR analysis confirmed that ECP broadly activates genes distinctive of monocyte maturation to DCs. Study of negatively enriched ECP-processed monocytes from 4 normal donors, to 95% population purity, confirmed the microarray finding that ECP rapidly initiates monocyte entry into the DC pathway. Expression of each of 11 examined DC-distinctive genes was significantly enhanced by ECP processing, 3 and > 1000-fold over that manifested by pretreatment monocytes. Nearly indistinguishable findings from identical analysis of nonpurified mononuclear populations indicates that ECP-lethally damaged lymphocytes do not discernibly contribute to the observed expression of these genes, even when present in the study population.
Discussion
This report provides new insights into the mechanistic basis of ECP's therapeutic efficacy and supports the premise that externally manipulated DCs can meaningfully contribute to immunotherapy of established serious disease. Whereas our prior results had suggested that ECP induces CTCL monocytes to differentiate into DCs, the data were not definitive for several reasons.
Definitive evidence of ECP's induction of monocyte-to-DC maturation requires demonstration of relevant gene activation. If normal monocytes should be similarly affected, ECP can be a general source of maturationally synchronized DCs. ECP-processed monocytes must remain sufficiently viable and healthy to selectively up-regulate the expression of genes relevant to DC immunologic functions and maturation and are substantially more resistant to the apoptotic influence of the treatment, in contrast to lymphocytes, which are universally susceptible to this effect. Finally, if ECP orchestrates predictable monocyte-to-DC maturation, it should induce a distinctive and reproducible transcriptome. This paper provides all of this requisite information.
The rapid generation of large numbers of functional new DCs, without the conventional approach of adding superpharmacologic concentrations of cytokines over 5-7 days of in vitro tissue culture, suggests that ECP induction of DCs may contribute to the procedure's recognized immunotherapeutic efficacy. Because DCs normally comprise less than 1% of blood mononuclear cells,11-13 ECP's efficient initiation of maturation of processed monocytes to DCs suggests that a large percentage of blood monocytes can be induced to differentiate into DCs.
Because they are so rapidly formed, the ECP-derived DCs are maturationally synchronized, as revealed by the enhanced relatively homogeneous level of expressed costimulatory surface molecules, such as CD86 (B7.2), when compared with DCs prepared in the conventional manner (cultured for 6 days with cytokines). This observation offers a potential explanation for how the same therapy can be both immunostimulatory in the setting of hematologic malignancy and immunosuppressive in the context of GVHD and organ transplant. Low expression of costimulatory molecules by immature DCs contributes to selective down-regulation of T-cell responses, whereas higher expression of the same membrane components can stimulate positive immunity.11-13
ECP was originally designed to enable the extracorporeal photoactivation of the DNA-cross-linking drug, 8-MOP, while blood is passaged as a 1-mm film through the ECP plastic ultraviolet exposure plate.2 We have previously shown that the passage through the plastic plate is critical for monocyte acquisition of the DC phenotype,7 because leukapheresed monocytes incubated overnight only minimally differentiate to DCs.
In the current study, we report that the large majority of the observed ECP-induced gene activation also requires passage through the plate and is first manifest after overnight incubation of the treated leukocytes. Now that the central importance of the exposure plate in ECP's induction of DCs is recognized, we conclude that experimental animal systems, which we and others have conceived and tested,6,33-35 lacking such an exposure plate, are not genuine ECP models.
Three factors appear to be required for the DC induction: (1) adherence of plasma proteins to the plastic UVA exposure plate, (2) passage of monocytes through the plate, and (3) overnight coincubation of the processed monocytes with 8-MOP–induced apoptotic lymphocytes. The manner in which these sequential influences complement each other and contribute to the DC induction and the specific roles of particular plasma proteins, after adherence to the exposure plate, can now be investigated.
These collective findings are compatible with the possibility that ECP's efficacy in immunocompetent CTCL patients may result, at least in part, from the loading of tumor antigens derived from apoptotic malignant cells into newly formed DCs, which then present them to a responding immune system. If that is the case, it may become possible to tailor ECP to treatment of other hematologic malignancies, as well as solid tumors. For example, apoptosis could be induced in malignant cells extracted from surgically excised tumors and then fed to similarly induced autologous DCs, which might then stimulate enhanced antitumor T-cell responses. Furthermore, the ease and rapidity with which ECP produces large numbers of synchronized DCs raises the possibility that ECP, appropriately modified by future advances, could become an attractive source of APCs for other DC-based protocols.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health, National Cancer Institute Spore grant 1 P50 CA121974 (C.B., F.F., M.G.), National Institutes of Health Cancer Center Support grant 3 P30 CA 16 359-28S1 (R.E.), the NY Cardiac Foundation (C.B., R.E.), and the Doris Duke Foundation (A.B.).
National Institutes of Health
Authorship
Contribution: C.B. performed experiments, directed research, analyzed data, and composed broad sections of the paper; J.L. and R.F. designed and conducted modified monocyte purification and RT-PCR analyses of DC gene expression in lymphocyte-depleted versus undepleted monocyte preparations; K.H., J.G.V., and S.M. performed gene expression experiments and contributed relevant portions of the manuscript; A.L., H.Z., and W.L. performed complex statistical analysis in the paper, including the principal component analysis, and contributed to the writing of those portions of the paper; T.D. and A.B. analyzed data; I.C. provided patient blood samples and processed the normal control blood in the ECP apparatus; F.F., M.G., and R.T. performed critical roles in experimental design and contributed to overall composition of the manuscript; and R.E. was involved in all phases of the design of the experiments, data interpretation, and writing of the manuscript.
Conflict-of-interest disclosure: C.B., R.E., R.T., and M.G. are listed as coinventors on Yale University-owned patents relating to monocyte-to-dendritic cell maturation. These patents have not been commercialized. The remaining authors declare no competing financial interests.
Correspondence: Carole Berger, Yale University School of Medicine, Department of Dermatology, 333 Cedar St, New Haven, CT 06520; e-mail: carole.berger@yale.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal