

Abstract
The prothrombinase complex converts prothrombin to α-thrombin through the intermediate meizothrombin (Mz-IIa). Both α-thrombin and Mz-IIa catalyze factor (F) XI activation to FXIa, which sustains α-thrombin production through activation of FIX. The interaction with FXI is thought to involve thrombin anion binding exosite (ABE) I. α-Thrombin can undergo additional proteolysis to β-thrombin and γ-thrombin, neither of which have an intact ABE I. In a purified protein system, FXI is activated by β-thrombin or γ-thrombin, and by α-thrombin in the presence of the ABE I-blocking peptide hirugen, indicating that a fully formed ABE I is not absolutely required for FXI activation. In a FXI-dependent plasma thrombin generation assay, β-thrombin, γ-thrombin, and α-thrombins with mutations in ABE I are approximately 2-fold more potent initiators of thrombin generation than α-thrombin or Mz-IIa, possibly because fibrinogen, which binds to ABE I, competes poorly with FXI for forms of thrombin lacking ABE I. In addition, FXIa can activate factor FXII, which could contribute to thrombin generation through FXIIa-mediated FXI activation. The data indicate that forms of thrombin other than α-thrombin contribute directly to feedback activation of FXI in plasma and suggest that FXIa may provide a link between tissue factor-initiated coagulation and the proteases of the contact system.
Introduction
The trypsin-like protease α-thrombin (α-IIa) is a key contributor to vital host responses to injury, including fibrin formation,1-3 platelet and endothelial cell activation,4 and inflammation.5 During coagulation, prothrombin is converted to α-IIa through a series of proteolytic steps catalyzed by factor (F) Xa.6,7 The process results in formation of a functional active site and expression of 2 anion binding exosites (ABE I and ABE II) that are required for α-IIa interactions with many substrates, receptors, and inhibitors (Table 1).1-3,6 In the presence of FVa and phospholipid, FXa initially converts prothrombin to the protease meizothrombin (Mz-IIa; Figure 1A),7-11 which expresses ABE I.6 Mz-IIa is rapidly converted to α-IIa, which may undergo further proteolysis to form β-thrombin (β-IIa) and γ-thrombin (γ-IIa) (Figure 1B), 2 proteases with reduced capacity to catalyze ABE I-dependent reactions.9-12 Physiologic roles for β-IIa or γ-IIa have not been established; however, both have been identified in clotting blood.11
Thrombin substrates and ligands1-3
| Thrombin substrate or binding partner | ABE I | ABE II | Comments |
|---|---|---|---|
| Fibrinogen | + | — | |
| Fibrinogen-γ′ | + | + | 10% of total fibrinogen contains γ′ chains |
| FV | + | + | IIa residues outside ABEs also involved |
| FVIII | + | + | IIa residues outside ABEs also involved |
| FXIII | — | — | Fibrin accelerates 80-fold |
| Protein C | — | — | Thrombomodulin facilitates interaction |
| TAFI | — | — | Thrombomodulin facilitates interaction |
| Heparin | — | + | |
| Antithrombin | — | — | Heparin facilitates interaction |
| Heparin cofactor II | + | — | Heparin facilitates interaction |
| Thrombomodulin | + | + | Glycosaminoglycans interact with ABE II |
| Glycoprotein 1bα | — | + | — |
| PAR-1 | + | — | — |
| PAR-4 | + | — | — |
| Thrombin substrate or binding partner | ABE I | ABE II | Comments |
|---|---|---|---|
| Fibrinogen | + | — | |
| Fibrinogen-γ′ | + | + | 10% of total fibrinogen contains γ′ chains |
| FV | + | + | IIa residues outside ABEs also involved |
| FVIII | + | + | IIa residues outside ABEs also involved |
| FXIII | — | — | Fibrin accelerates 80-fold |
| Protein C | — | — | Thrombomodulin facilitates interaction |
| TAFI | — | — | Thrombomodulin facilitates interaction |
| Heparin | — | + | |
| Antithrombin | — | — | Heparin facilitates interaction |
| Heparin cofactor II | + | — | Heparin facilitates interaction |
| Thrombomodulin | + | + | Glycosaminoglycans interact with ABE II |
| Glycoprotein 1bα | — | + | — |
| PAR-1 | + | — | — |
| PAR-4 | + | — | — |
+ indicates that ABE I or ABE II is required for the interaction; and —, an ABE interaction is not required.

Products of prothrombin activation. (A) Sodium dodecyl sulfate-polyacrylamide gel showing recombinant prothrombin (II) and recombinant prothrombin with alanine substitutions for Arg155, Arg271, and Arg284 (Mz). The latter has been converted to the active protease Mz-IIa by incubating with ecarin. The numbers to the right of the gel indicate the amino acids contained in the 2 major fragments of Mz-IIa. The schematic diagram to the right shows conversion of prothrombin to α-thrombin, with the intermediates Mz-IIa and Mz-IIa(desF1), and are labeled using a numbering system for human prothrombin. (B) Nonreducing SDS-polyacrylamide gel showing preparations of α-IIa, β-IIa, and γ-IIa, relative to prothrombin (II). In the schematic diagram to the right, the proteases are labeled using separate numbering systems for the human α-IIa A1′ and B chains. For α-IIa, the corresponding numbers for the prothrombin numbering system (top) are shown for comparison. Gels in both panels were stained with Coomassie blue, and the positions of molecular mass standards in kilodaltons are shown to the left of the gels.
Products of prothrombin activation. (A) Sodium dodecyl sulfate-polyacrylamide gel showing recombinant prothrombin (II) and recombinant prothrombin with alanine substitutions for Arg155, Arg271, and Arg284 (Mz). The latter has been converted to the active protease Mz-IIa by incubating with ecarin. The numbers to the right of the gel indicate the amino acids contained in the 2 major fragments of Mz-IIa. The schematic diagram to the right shows conversion of prothrombin to α-thrombin, with the intermediates Mz-IIa and Mz-IIa(desF1), and are labeled using a numbering system for human prothrombin. (B) Nonreducing SDS-polyacrylamide gel showing preparations of α-IIa, β-IIa, and γ-IIa, relative to prothrombin (II). In the schematic diagram to the right, the proteases are labeled using separate numbering systems for the human α-IIa A1′ and B chains. For α-IIa, the corresponding numbers for the prothrombin numbering system (top) are shown for comparison. Gels in both panels were stained with Coomassie blue, and the positions of molecular mass standards in kilodaltons are shown to the left of the gels.
α-IIa up-regulates its own generation by activating the cofactors FV and FVIII,1-3,9 and by converting FXI to the protease FXIa.13,14 FXIa, in turn, sustains α-IIa generation by converting FIX to FIXaβ,15 and possibly by activating FV and FVIII.16 In the original cascade/waterfall hypotheses of coagulation, FXI is activated by FXIIa17,18 ; however, current models deemphasize this reaction based on the observation that FXI deficiency is associated with abnormal bleeding, whereas FXII deficiency is not.18 FXI activation by α-IIa would explain the phenotypic differences between FXI and FXII deficiency.19,20 FXI is a homolog of prekallikrein (PK), the zymogen of a protease (α-kallikrein) involved in kinin formation.17 Although FXI and PK are both FXIIa substrates, PK is not activated by α-IIa.20 FXI has features that may facilitate activation by α-IIa, including sequence around the activation cleavage site typical of α-IIa substrates (Tables 2–3).9 Yun et al showed that amino acids in ABE I are required for optimal FXI activation by α-IIa.21 Mz-IIa also activates FXI,22 consistent with a role for ABE I in the interaction with FXI. As β-IIa and γ-IIa have reduced ABE I expression, it seems reasonable to postulate they would interact poorly with FXI; however, we show that β-IIa and γ-IIa do activate FXI and are more potent initiators of FXI-dependent thrombin generation in plasma than are α-IIa or Mz-IIa.
FXI and PK activation sites
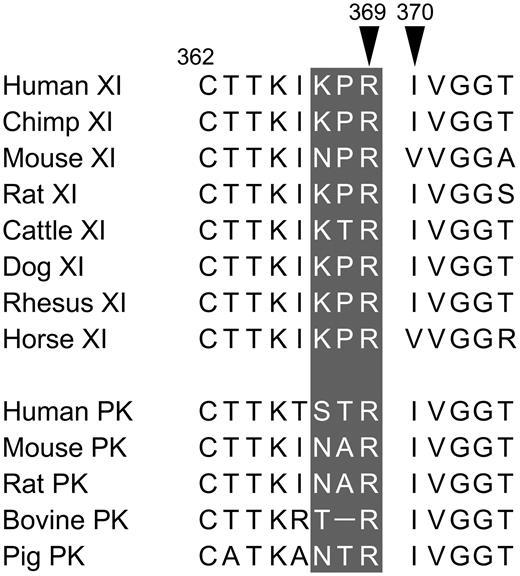
Shown are amino acid sequences around the FXI and PK cleavage sites. Positions of amino acids 369 (P1 residue) and 370 (P1′ residue) are shown relative to the conserved cysteine at position 362. The P3-P1 residues are shown in white on the gray background.
α-Thrombin cleavage sites

Shown are amino acid sequences around the cleavage sites for substrates of α-thrombin. Positions of the P1 and P1′ residues are indicated. The P3-P1 residues are shown in white on the gray background. For substrates with multiple cleavage sites, the number of the P1 position is indicated in parentheses. Data are from Jenny et al.9
Methods
Reagents
Reagents included the following: normal human plasma (Precision BioLogic), FXII-deficient plasma (George King Biomed), FXII, FXIIa, PK, α-kallikrein, γ-IIa, corn-trypsin inhibitor (CTI), and goat polyclonal IgG against human FXI, FXII, and PK (Enzyme Research Lab). γ-IIa was prepared by digestion of α-IIa with trypsin immobilized on Sepharose. Others included the following: FVIIa, FIXa, FXa, FXI, FXIa, α-IIa, β-IIa, Phe-Pro-Arg-chloromethylketone (FPR-CK), Phe-Phe-Arg-chloromethylketone (FFR-CK) (Haematologic Technologies). β-IIa was prepared by limited digestion of α-IIa with bovine trypsin using the method of Braun et al.23 Others included: Argatroban (GlaxoSmithKline), Lepirudin (hirudin) (Bayer), dextran sulfate (average molecular mass, 500 000 Da), hirugen, bovine serum albumin, bovine trypsin, Oxyuranus scutellatus venom, Echis carinatus venom (ecarin), rabbit brain cephalin, ovomucoid trypsin inhibitor (OTI), soybean trypsin inhibitor, and phenylmethylsulfonyl fluoride (Sigma-Aldrich); S-2366 (L-pyro-Glu-L-Pro-L-Arg-p-nitroanilide), S-2302 (H-D-Pro-L-Phe-L-Arg-p-nitroanilide, S-2238 (H-D-Phe-L-Pip-L-Arg-p-nitroanilide), DiaPharma. Z-Gly-Gly-Arg-AMC (Bachem); and phosphatidylcholine/phosphatidylserine (PC/PS) vesicles (Avanti Polar Lipids).
Recombinant protein
FXI.
Wild-type (WT) FXI and FXI with alanine replacing Ser557 (FXI-Ala557) were expressed in HEK293 cells and purified from conditioned media (Cellgro Complete, Mediatech) using anti–human FXI-IgG 1G5.12.24 FXI was dialyzed against 50mM tris(hydroxymethyl)aminomethane HCl, pH 7.4, 100mM NaCl (Tris-buffered saline [TBS]), and stored at −80°C.
Mz-IIa.
AD293 fibroblasts transfected with pcDNA3.1 containing a human prothrombin complementary deoxyribonucleic acid with alanine substitutions for Arg155, Arg271, and Arg284 were grown in Cellgro Complete Media with 10 μg/mL vitamin K1.8 Conditioned media supplemented with phenylmethylsulfonyl fluoride, FPR-CK, FFR-CK (0.1mM), and benzamidine (1mM) was applied to a Q-Sepharose Fast Flow column (Amersham) equilibrated with 20mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 250mM NaCl, 1mM benzamidine, 4mM ethylenediaminetetraacetic acid, pH 7.4, and eluted by 0 to 20mM CaCl2 gradient. Fractions containing prothrombin were dialyzed against 10mM potassium phosphate, pH 7.5, 0.1μM FPR-CK/FFR-CK, and applied to a CHT ceramic hydroxyapatite type I column (Bio-Rad). Protein was eluted with 10 to 500mM potassium phosphate, pH 7.5. Prothrombin (20μM) was mixed with ecarin (2 units/mL) in 10 mL of 100mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 100mM NaCl, 5mM CaCl2, 1 mg/mL polyethylene glycol 8000, pH 7.4. After incubation at room temperature for 4.5 hours, the reaction was quenched with 20mM ethylenediaminetetraacetic acid, diluted 5-fold in 20mM sodium citrate, pH 6.0, and applied to a Resource Q column (Amersham). rMz-IIa was eluted with a 0 to 1M NaCl gradient in 20mM sodium citrate, pH 6.0. Protein was dialyzed against 50mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 110mM NaCl, 5mM CaCl2, pH 7.4, and stored at −80°C.
α-IIa.
WT prothrombin and prothrombin with Arg68 to Glu68 or Arg70 to Glu70 substitutions (α-IIa B chain numbering, Figure 1B) were prepared as described.25,26 Protein expressed in CV-1 cells was partially purified from media by barium sulfate precipitation and chromatography on DEAE-Sephacel. α-IIa was generated by incubating at 37°C for 60 minutes with 0.01 volume of 2.8 mg/mL Oxyuranus scutellatus venom preabsorbed with Amberlite CG-50 in buffer containing 0.01 volume rabbit brain cephalin and 5mM CaCl2. α-IIa-WT α-IIa-Glu68, and α-IIa-Glu70 were purified by Amberlite CG-50 chromatography. Concentration was determined by active site titration with hirudin.26
Plasma clotting assay
Active site concentrations for Mz-IIa, β-IIa, and γ-IIa were determined by amidolytic assay using substrate S-2238. OTI was added to γ-IIa to inhibit contaminating trypsin. Protein concentrations were normalized to an α-IIa preparation in which active site concentration was determined by hirudin titration. Clotting times were determined on an ST-4 Coagulation Analyzer (Diagnostica Stago). Thrombin or trypsin was diluted in Tyrode buffer (15mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.4, 125mM NaCl, 2.7mM KCl, 1mM MgCl2, 0.4mM NaH2PO4, 5.6mM dextrose, 0.35% BSA), and 10 μL was mixed with 80 μL Tyrode buffer with or without 10mM CaCl2. Reactions were initiated with 30 μL normal plasma. Before use, plasmas were incubated for 30 minutes at 4°C with vehicle, antibodies O1A6 or 14E11 (300nM),19 or CTI (4μM). A clotting time more than 600 seconds was considered to indicate no activity.
Activation of FXI by thrombin
Reactions were performed in TBS, 0.1% PEG 8000. FXI-WT (100nM) was incubated with thrombin (25nM) at 37°C. At various time points, 50 μL aliquots were supplemented with argatroban (5μM) and added to 50 μL 1mM S-2366. Change in absorbance at OD 405 nm was followed on a SpectraMAX 340 microtiter plate reader (Molecular Devices). Values were converted to nanomolar FXIa using a standard curve. Effects of FV or FVa on FXI activation by thrombin were determined by the method of Maas et al.27 Briefly, FXI (30nM) was incubated with thrombin (10nM) with or without FV or FVa (30nM, a gift from Dr Paul Bock) in Tyrode buffer with 3mM CaCl2 at 37°C for 10 minutes. Argatroban was added to 5μM, and FXIa generation determined by monitoring cleavage of S-2366. FXI activation was also studied by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. FXI-Ala557 (100-200nM) and proteases (25-50nM) were incubated in TBS, 30μM ZnCl2, 0.1% PEG 8000 at 37°C. Reactions with dextran sulfate were done in TBS, 1 μg/mL dextran sulfate, 0.1% PEG 8000 at 37°C, using 60nM FXI-Ala557 and 2nM α-IIa. At various time points, aliquots were removed into reducing sample buffer, size fractionated by SDS-PAGE, and analyzed by Western blot with goat polyclonal anti–human FXI IgG.
Thrombin generation assay
Thrombin generation in plasma was measured by following cleavage of Z-Gly-Gly-Arg-AMC at 37°C on a thrombinoscope.19 Before testing, normal plasma underwent centrifugation at 13 000g for 30 minutes to remove chylomicrons (CMs). Thrombin generation assays (TGAs) were performed in 96-well round bottom plates coated with PEG 20 000. Plasma was supplemented with 415μM Z-Gly-Gly-Arg-AMC, 5μM PC/PS vesicles, 4μM CTI or vehicle, and 50 μg/mL O1A6 or vehicle. Supplemented plasma (40 μL) was mixed with 10 μL Tyrode buffer containing thrombin or trypsin (10nM final concentration). In some reactions, hirugen (10μM) was included; 10 μL of 20mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.4, 100mM CaCl2, 6% BSA was added and fluorescence (excitation λ 390 nm, emission λ 460 nm) was monitored. Each set of conditions was run 3 times in duplicates. Peak thrombin generation and area under the thrombin generation curve (endogenous thrombin potential [ETP]) were calculated using Thrombinoscope Analysis software, Version 3.0.
Activation of FXII and PK
FXII autoactivation was studied in TBS, 30μM ZnCl2, 0.1% PEG 8000, and 0.5mM S-2302 in the presence of CMs (a gift from Dr Macrae Linton), or PC/PS vesicles at room temperature. Changes in OD 405 nm were followed on the SpectraMAX 340 plate reader. FXII or PK activation by FXIa was tested as follows: FXII or PK (200nM) was incubated with or without FXIa (25nM) in TBS, 0.1% PEG 8000 at 37°C. For FXII, at various times, 90 μL of reaction was mixed with SBTI (500nM) and S-2302 (0.5mM), and changes in OD 405 nm were followed. Reactions with PK were treated similarly, except that FXIa was preincubated with CTI to block FXIIa, and no SBTI was used. For SDS-PAGE analysis, FXII or PK (200nM) was incubated in TBS, 0.1% PEG 8000 with coagulation proteases (25nM) or trypsin (0.1nM) at 37°C. Reactions were stopped with reducing sample buffer, size-fractionated on 10% polyacrylamide gels, and analyzed by Western blot using polyclonal IgG against the relevant protein.
Results
FXI cleavage by coagulation proteases
FXI can undergo autocatalytic activation that is accelerated by polyanions, such as dextran sulfate.13,14 In FXI-Ala557 (Figure 2A), the active site serine is replaced with alanine. FXIIa converts FXI-WT and FXI-Ala557 to FXIa and FXIa-Ala557 by cleaving the Arg369-Ile370 bond on each subunit. Unlike FXIaWT, FXIa-Ala557 lacks measurable catalytic activity and can be used to assess FXI activation without contribution from autoactivation. Conversion of the 80-kDa FXI zymogen subunit to the 50- and 30-kDa heavy and light chains of FXIa is followed by reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Figure 2B shows FXI-Ala557 incubated with various proteases (4:1 substrate/enzyme). Activation is evident with α-IIa and FXIIa, but not α-kallikrein, FIXaβ, FXa, or FVIIa/tissue factor. The P3 to P1 residues preceding the activation cleavage site in human FXI (Lys-Pro-Arg369)28 are conserved in most mammals (Table 2) and differ from corresponding residues in the homolog PK.29

Recombinant FXI-Ala557. (A) Shown are nonreducing (left panel) and reducing (right panel) sodium dodecyl sulfate-polyacrylamide gels of human FXI from plasma (pXI), recombinant WT FXI, and FXI with an alanine substitution for Ser557. FXI is a disulfide-linked homodimer, which accounts for the decrease in apparent molecular mass on reduction. (B) Western blot of FXI-Ala557 (200nM) incubated for 1 hour with 50nM α-IIa (IIa), α-kallikrein (Ka), FXIIa (XIIa), FIXaβ (IXa), FXa (Xa), or FVIIa with tissue factor (VIIa). The position of the zymogen FXI band (Z) and the heavy chain (HC) and light chain (LC) of FXIa are shown at the right of the gel. Lanes labeled XI and XIa contain samples of purified FXI and FXIa, respectively. The blot was developed with a goat polyclonal anti–human FXI antibody. For both panels, positions of molecular mass standards in kilodaltons are shown to the left of the gels.
Recombinant FXI-Ala557. (A) Shown are nonreducing (left panel) and reducing (right panel) sodium dodecyl sulfate-polyacrylamide gels of human FXI from plasma (pXI), recombinant WT FXI, and FXI with an alanine substitution for Ser557. FXI is a disulfide-linked homodimer, which accounts for the decrease in apparent molecular mass on reduction. (B) Western blot of FXI-Ala557 (200nM) incubated for 1 hour with 50nM α-IIa (IIa), α-kallikrein (Ka), FXIIa (XIIa), FIXaβ (IXa), FXa (Xa), or FVIIa with tissue factor (VIIa). The position of the zymogen FXI band (Z) and the heavy chain (HC) and light chain (LC) of FXIa are shown at the right of the gel. Lanes labeled XI and XIa contain samples of purified FXI and FXIa, respectively. The blot was developed with a goat polyclonal anti–human FXI antibody. For both panels, positions of molecular mass standards in kilodaltons are shown to the left of the gels.
Thrombin preparations and thrombin time clotting assays
Proteases derived from prothrombin are shown in Figure 1. As plasma Mz-IIa rapidly converts to α-IIa, we prepared a recombinant stable form of the protease (rMz-IIa, Figure 1A).8,30 β-IIa and γ-IIa are cleaved at the Arg70-Tyr71 bond (numbering from the α-IIa B-chain N-terminus), and γ-IIa has an additional cleavage at the Lys154-Gly155 bond (Figure 1B). β-IIa may contain a trace of α-IIa, and a product similar (but not identical) to γ-IIa. γ-IIa may contain a small amount of β-IIa. β-IIa and γ-IIa were prepared by limited digestion of α-IIa with trypsin. By Western blot, a trace of trypsin was detected in γ-IIa (∼ 0.05% molar concentration) and was not detected in β-IIa. OTI, a protein isolated from egg white, is an effective trypsin inhibitor but had no detectable effect on α-IIa, β-IIa, γ-IIa, FXIa, FXIIa, or α-kallikrein in chromogenic substrate assays, and does not affect plasma clotting time (data not shown). Plasma-derived thrombins were preincubated with OTI in some studies to determine whether trypsin was affecting results.
The capacity of α-IIa (specific activity ∼ 2800 units/mg) to induce fibrin formation in normal plasma anticoagulated with sodium citrate is shown in Figure 3A. Consistent with published results,9,10,12 rMz-IIa and β-IIa exhibited approximately 10% and 3% of the fibrinogen clotting activity of α-IIa, respectively, whereas γ-IIa did not have measurable activity (Figure 3B). This assay assesses the capacity of the added thrombin to directly convert fibrinogen to fibrin, as the low free calcium concentration in citrate-treated plasma limits α-IIa formation from endogenous prothrombin. The data support the premise that ABE I is not in a fully active conformation in β-IIa or γ-IIa. ABE I is actually formed in Mz-IIa6,31 ; however, activity toward fibrinogen is decreased compared with α-IIa because of an effect on catalysis (decreased kcat), and not a lower affinity (increased Km) for fibrinogen.32

Plasma thrombin clotting assay. (A) Time to clot formation after addition of various concentrations of α-thrombin (listed at bottom) to pooled normal plasma collected into 0.32% sodium citrate in the absence of recalcification. The column at the extreme left shows results for plasma in the absence of recalcification or added thrombin, whereas the second column from the left shows that plasma does not clot after recalcification in the absence of added thrombin. (B) Time to clot formation after addition of various concentrations of Mz-IIa, β-IIa, or γ-IIa to pooled normal plasma collected into 0.32% sodium citrate in the absence of recalcification. Forms and final concentrations indicated at the bottom of the panel. (C-F) Time to clot formation after addition of (C) 1.25nM α-IIa, (D) 2.5nM rMz-IIa, (E) 5nM β-IIa, or (F) 10nM γ-IIa to pooled normal plasma collected into 0.32% sodium citrate, with (+Ca2+) or without (−Ca2+) recalcification. Recalcified reactions were run in the presence of vehicle (V), an IgG that blocks FIX activation by FXIa (O1A6), an IgG that blocks FXI activation by FXIIa (14E11), or a FXIIa inhibitor (CTI). For panel F, reactions are also run in the presence of ovomucoid trypsin inhibitor (OTI). For all panels, each symbol indicates a single clotting time, and bars indicate the mean clotting time for each group.
Plasma thrombin clotting assay. (A) Time to clot formation after addition of various concentrations of α-thrombin (listed at bottom) to pooled normal plasma collected into 0.32% sodium citrate in the absence of recalcification. The column at the extreme left shows results for plasma in the absence of recalcification or added thrombin, whereas the second column from the left shows that plasma does not clot after recalcification in the absence of added thrombin. (B) Time to clot formation after addition of various concentrations of Mz-IIa, β-IIa, or γ-IIa to pooled normal plasma collected into 0.32% sodium citrate in the absence of recalcification. Forms and final concentrations indicated at the bottom of the panel. (C-F) Time to clot formation after addition of (C) 1.25nM α-IIa, (D) 2.5nM rMz-IIa, (E) 5nM β-IIa, or (F) 10nM γ-IIa to pooled normal plasma collected into 0.32% sodium citrate, with (+Ca2+) or without (−Ca2+) recalcification. Recalcified reactions were run in the presence of vehicle (V), an IgG that blocks FIX activation by FXIa (O1A6), an IgG that blocks FXI activation by FXIIa (14E11), or a FXIIa inhibitor (CTI). For panel F, reactions are also run in the presence of ovomucoid trypsin inhibitor (OTI). For all panels, each symbol indicates a single clotting time, and bars indicate the mean clotting time for each group.
Recalcifying plasma in the absence of thrombin did not induce clotting during the observation period (Figure 3A second column from left) and had little effect on the relatively short clotting times with 1.25nM α-IIa (Figure 3C). With α-IIa, results were not affected by antibodies that block FIX activation by FXIa (O1A6)19 or FXI activation by FXIIa (14E11),19,33 or by a FXIIa inhibitor (CTI), indicating any FXIa or FXIIa formed does not influence fibrin formation. rMz-IIa at 2.5nM induced clotting with recalcification, but not without it, indicating that α-IIa must be forming from endogenous prothrombin (Figure 3D). Again, this does not require FXIa or FXIIa. The mechanism by which rMz-IIa facilitates α-IIa formation here is not clear but may involve FV activation. β-IIa (Figure 3E) and γ-IIa (Figure 3F) at 5 and 10nM, respectively, induced clotting only in recalcified plasma, consistent with generation of α-IIa from prothrombin. O1A6 had a modest effect with β-IIa and blocked clotting with γ-IIa, suggesting that these proteases activate FXI in plasma. The result cannot be attributed to trypsin contamination of γ-IIa, as OTI did not affect results (Figure 3F), and 2.5nM bovine trypsin (∼ 500-fold greater than estimated contamination of γ-IIa) does not induce clotting (not shown). These data show that β-IIa and γ-IIa facilitate α-IIa generation through a process involving FXI and suggest that β-IIa and γ-IIa activate FXI. Previously, we showed that α-IIa activates FXI in plasma.19 However, this would not be evident in an assay using time to clot formation as an endpoint because α-IIa is an efficient inducer of fibrin formation and the bulk of FXI-dependent α-IIa generation occurs after the clot forms. β-IIa and γ-IIa, in contrast, convert fibrinogen to fibrin poorly, allowing FXI activation and activity to become an important determinant of time to clot formation.
FXI activation by plasma-derived thrombin
Published work with α-IIa variants indicate ABE I is involved in the FXI-α-IIa interaction.21 Alanine substitutions at residues 68, 70, 71, or 73 (α-IIa B-chain numbering system) reduced activity toward FXI.21 The effect of thrombin on FXI was studied by Western blot. As expected, FXI-Ala557 was readily converted to FXIa-Ala557 by α-IIa (Figure 4A), but β-IIa and γ-IIa also catalyzed FXI-Ala557 cleavage, although less efficiently than α-IIa (Figure 4A). In a FXIa chromogenic substrate assay, α-IIa is an approximately 2- to 3-fold better activator of FXI than β-IIa and approximately 4- to 5-fold better than γ-IIa (Figure 4B), supporting the premise that ABE I is required for optimal FXI activation.21 A recent report suggests that FV modestly enhances FXI activation by α-IIa.27 We tested both FV and FVa in the chromogenic substrate assay but saw no evidence for an effect on FXI activation with any type of thrombin.

FXI activation by thrombin. (A) Western blot of FXI-Ala557 (100nM) incubated with 25nM α-IIa, β-IIa, or γ-IIa, or 0.1nM bovine trypsin. FXI was detected with a polyclonal anti–human FXI antibody. Incubation times in hours are indicated below the blot. The position of the zymogen FXI band (Z) and the heavy chain (HC) and light chain (LC) of FXIa are shown to the left of the gel. (B) FXI-WT (100nM) was incubated with 25nM α-IIa (■), β-IIa (□), γ-IIa (●), or vehicle (○) at 37°C. FXIa generation was determine with a chromogenic substrate assay on aliquots at various time points as described in “Activation of FXI by thrombin.” (C-F) Thrombin generation assays were run as described in “Thrombin generation assay.” (C) Thrombin generation initiated by 10nM α-IIa, β-IIa, or γ-IIa in FXII-deficient plasma #1. (D) Same as in panel C, but with FXII-deficient plasma #2. (E) Same as in panel C with the addition of 300nM IgG O1A6 to block FIX activation by FXIa. (F) FXII-deficient plasma #1 using 10nM trypsin as the initiator. The signal here is probably background from trypsin cleavage of the fluorogenic substrate.
FXI activation by thrombin. (A) Western blot of FXI-Ala557 (100nM) incubated with 25nM α-IIa, β-IIa, or γ-IIa, or 0.1nM bovine trypsin. FXI was detected with a polyclonal anti–human FXI antibody. Incubation times in hours are indicated below the blot. The position of the zymogen FXI band (Z) and the heavy chain (HC) and light chain (LC) of FXIa are shown to the left of the gel. (B) FXI-WT (100nM) was incubated with 25nM α-IIa (■), β-IIa (□), γ-IIa (●), or vehicle (○) at 37°C. FXIa generation was determine with a chromogenic substrate assay on aliquots at various time points as described in “Activation of FXI by thrombin.” (C-F) Thrombin generation assays were run as described in “Thrombin generation assay.” (C) Thrombin generation initiated by 10nM α-IIa, β-IIa, or γ-IIa in FXII-deficient plasma #1. (D) Same as in panel C, but with FXII-deficient plasma #2. (E) Same as in panel C with the addition of 300nM IgG O1A6 to block FIX activation by FXIa. (F) FXII-deficient plasma #1 using 10nM trypsin as the initiator. The signal here is probably background from trypsin cleavage of the fluorogenic substrate.
Previously, we described an FXII-deficient plasma system in which thrombin generation from endogenous prothrombin requires FXI, Ca2+, and a trigger, such as a small amount of α-IIa.19 Results supported the conclusion that α-IIa activates FXI in plasma. Figure 4C-D shows results with this system for 2 FXII-deficient plasmas. Thrombin generation was initiated with 10nM α-, β-, or γ-IIa. β-IIa and γ-IIa were significantly better triggers than α-IIa in both plasmas, with more than 2-fold higher peak thrombin generation and 1.5- to 2-fold greater ETP (Figure 4C, 238, 526, and 487 nM·min; Figure 4D, 529, 810, and 901 nM·min for α-, β-, and γ-IIa, respectively). In all cases, the process was FXI-dependent, as O1A6 blocked thrombin generation (Figure 4E). Trypsin (10nM) did not induce thrombin generation (Figure 4F). These data show that β-IIa and γ-IIa are better inducers of FXI-dependent thrombin generation than α-IIa in plasma and strongly suggest that the proteases are more efficient activators of FXI in this environment, despite their lower activity in the purified protein system. This could reflect reduced competition from fibrinogen for binding to β-IIa and γ-IIa because of loss of ABE I expression.
The polyanion dextran sulfate markedly enhances FXI activation by α-IIa or autoactivation.13,14,20 The effect of dextran sulfate on cleavage of FXI-Ala557 (which does not autoactivate) by thrombin is shown in Figure 5A. In these Western blots, the FXIa light chain did not stain well, which is an occasional problem with the primary antibody we use. The heavy chain, however, is clearly visible. Hirudin, an α-IIa inhibitor from leech saliva, blocks the α-IIa active site and ABE I and, not surprisingly, is a potent inhibitor of FXI activation by α-IIa (Figure 5B). In contrast, hirugen (a peptide from hirudin that blocks ABE I, but not the active site) did not inhibit FXI-Ala557 cleavage (Figure 5C), strongly indicating that FXI does not need to interact with ABE I to be activated by α-IIa. The reported low activities of α-IIa with ABE I mutations toward FXI may, therefore, not be directly the result of loss of ABE I activity.21 We postulated that adding hirugen to the TGA would enhance FXI-dependent thrombin generation by interfering with fibrinogen binding to ABE I. This would make α-IIa more available to FXI. Indeed, hirugen increased α-IIa-induced thrombin generation approximately 4-fold (Figure 5D, 579 and 2148 nM·min). This study used twice the reaction volume of studies in Figure 4, accounting for the larger baseline result. Taken as a whole, the data indicate that loss of ABE I expression in β-IIa and γ-IIa, although associated with a reduction in FXI activation in a purified system, actually facilitates FXI activation by these proteases in a plasma environment replete with proteins that bind ABE I on α-IIa.

Effect of hirugen on FXI activation by α-IIa. (A-C) Western blots of FXI-Ala557 (60nM) incubated with 2nM α-IIa in the presence of 1 μg/mL dextran sulfate and (A) vehicle, (B) hirudin (12 units/mL), or (C) hirugen (10μM). The blot was developed with a goat polyclonal antihuman FXI antibody. Positions of zymogen FXI (Z) and the heavy chain (HC) and light chain (LC) of FXIa are shown to the right of the blots. (D) Thrombin generation assay. Thrombin generation was initiated in FXII-deficient plasma with vehicle (−IIa) or 10nM α-IIa in the absence (−hirugen) or presence (+hirugen) of 10μM hirugen. The volumes used here are twice as large as those in Figure 4.
Effect of hirugen on FXI activation by α-IIa. (A-C) Western blots of FXI-Ala557 (60nM) incubated with 2nM α-IIa in the presence of 1 μg/mL dextran sulfate and (A) vehicle, (B) hirudin (12 units/mL), or (C) hirugen (10μM). The blot was developed with a goat polyclonal antihuman FXI antibody. Positions of zymogen FXI (Z) and the heavy chain (HC) and light chain (LC) of FXIa are shown to the right of the blots. (D) Thrombin generation assay. Thrombin generation was initiated in FXII-deficient plasma with vehicle (−IIa) or 10nM α-IIa in the absence (−hirugen) or presence (+hirugen) of 10μM hirugen. The volumes used here are twice as large as those in Figure 4.
FXI activation by recombinant thrombin
Cleavage of FXI-Ala557 by recombinant thrombin was studied with Western blots. α-IIa-WT readily converts FXI-Ala557 to FXIa-Ala557 (Figure 6A-B), whereas rMz-IIa cleaved FXI-Ala557 approximately 4-fold more rapidly than α-IIa (Figure 6C). The ABE I variant α-IIa-Glu68, which cleaves fibrinogen poorly (< 1% α-IIa-WT activity),26 exhibited several-fold reduced activity toward FXI (Figure 6D), consistent with published results.21 α-IIa-Glu70, which activates protein C poorly in the presence of thrombomodulin,25 activated FXI comparably to α-IIa. In the TGA, thrombin generation induced by α-IIa-WT (Figure 6F, ETP 255 nM·min) was similar to plasma α-IIa (Figure 4C, ETP 238 nM·min), but rMz-IIa was slightly less active than α-IIa-WT (Figure 6F, ETP 185 nM·min). Thrombin generation was approximately 2-fold greater with α-IIa-Glu68 and α-IIa-Glu70 (556 and 548 nM·min, respectively) than with α-IIa-WT (Figure 6G). Again, the data suggest that α-IIa species with reduced ABE I expression have an advantage in plasma over α-IIa-WT or rMz-IIa because of reduced competition from plasma components that bind to residues in ABE I.

FXI activation by recombinant thrombins. (A-E) Western blot of FXI-Ala557 (100nM) incubated with (A) vehicle, or 25nM (B) α-IIa-WT, (C) rMz-IIa, (D) α-IIa-Glu68, or (E) α-IIa-Glu70 at 37°C. FXI was detected with a polyclonal goat antihuman FXI antibody. Positions of zymogen FXI (Z) and the heavy chain (HC) and light chain (LC) of FXIa are shown to the left of the blots. Incubation times in hours are indicated above panel A. (F-G) Thrombin generation initiated in FXII-deficient plasma by 10nM (F) α-IIa-WT or rMz-IIa, or (G) α-IIa-Glu68 and α-IIa-Glu70.
FXI activation by recombinant thrombins. (A-E) Western blot of FXI-Ala557 (100nM) incubated with (A) vehicle, or 25nM (B) α-IIa-WT, (C) rMz-IIa, (D) α-IIa-Glu68, or (E) α-IIa-Glu70 at 37°C. FXI was detected with a polyclonal goat antihuman FXI antibody. Positions of zymogen FXI (Z) and the heavy chain (HC) and light chain (LC) of FXIa are shown to the left of the blots. Incubation times in hours are indicated above panel A. (F-G) Thrombin generation initiated in FXII-deficient plasma by 10nM (F) α-IIa-WT or rMz-IIa, or (G) α-IIa-Glu68 and α-IIa-Glu70.
FXI-dependent thrombin generation in the presence of FXII
When TGAs are run with normal plasma, a FXIIa inhibitor, such as CTI, is usually included. In the absence of CTI, thrombin generation frequently occurs in the absence of a trigger. This is attributed to FXII activation during phlebotomy or plasma preparation and is considered an undesirable artifact. We also observe a variable tendency for spontaneous thrombin generation to occur in normal plasma that can be blocked with CTI or O1A6, consistent with FXIIa activation of FXI. However, centrifugation of plasma to remove low-density CMs prevents spontaneous thrombin generation, whereas adding CMs back to plasma depleted of CMs restores spontaneous thrombin generation (not shown). Purified CMs stimulated FXII autoactivation (Figure 7A), whereas the PC/PS vesicles used in the TGA were relatively weak stimulators. To study the effects of α-, β-, and γ-IIa on thrombin generation in the presence of FXII without CTI, TGAs were run using normal plasma depleted of CMs. Spontaneous thrombin generation did not occur in this plasma, but FXI-dependent thrombin generation was robust when a 10nM thrombin trigger was used (Figure 7B-D). As in FXII-deficient plasma (Figure 4C), thrombin generation in normal plasma triggered with α-IIa (Figure 7B, ETP 663 nM·min) was less than with β-IIa (Figure 7C, ETP 966 nM·min) or γ-IIa (Figure 7D, ETP 930 nM·min). Surprisingly, addition of CTI reduced thrombin generation by more than half for all initiators (ETPs, 302, 455, and 338 nM·min, for α-, β-, or γ-IIa, respectively), indicating that FXII was activated during the reactions. We postulated that FXII activation could be mediated directly by α-, β-, or γ-IIa, or indirectly through conversion of PK to the potent FXII activator α-kallikrein. However, neither PK (Figure 7E) nor FXII (Figure 7F) is cleaved appreciably by α-, β-, or γ-IIa. However, FXIa did activate FXII (Figure 7F), but not PK (Figure 7E). This was confirmed with chromogenic substrate assays for α-kallikrein (Figure 7G) and FXIIa (Figure 7H). These data raise the possibility that FXIa generated by thrombin can activate FXII. The resulting FXIIa could then contribute to thrombin generation through activation of additional FXI or activate the kallikrein-kinin system through activation of PK.

FXI activation in normal plasma. (A) Autoactivation of FXII in the presence of 50nM PC/PS vesicles, 50nM CMs, or vehicle control. Reactions were run as described in “Activation of FXII and PK.” (B-D) Thrombin generation in normal plasma depleted of CMs (“Thrombin generation assay”) initiated by 10nM (B) α-IIa, (C) β-IIa, or (D) γ-IIa in the presence or absence of 4μM CTI, or the presence of 300 μg/mL O1A6. (E-F) Western blots of (E) PK or (F) FXII (200nM) incubated with vehicle (C) or 25nM α-IIa (α), β-IIa (β), γ-IIa (γ), FXIa (XIa), FXIIa (XIIa), α-kallikrein (Ka), or 0.01nM trypsin (T). Blots were developed with goat polyclonal antibodies against the relevant protein. α-Kallikrein and FXIIa standards (St) are in the first lanes of each blot, with positions of zymogen PK and FXII (Z) and the heavy chain (HC) and light chain (LC) of α-kallikrein and FXIIa shown to the left of each blot. β-kal indicates bands representing the degradation product β-kallikrein. (G-H) Chromogenic substrate assays for activation of (G) PK or (H) FXII (200nM) by 25nM FXIa (●) or vehicle (□) at 37°C. α-Kallikrein or FXIIa generation was assessed by chromogenic substrate assay as described in “Activation of FXII and PK.”
FXI activation in normal plasma. (A) Autoactivation of FXII in the presence of 50nM PC/PS vesicles, 50nM CMs, or vehicle control. Reactions were run as described in “Activation of FXII and PK.” (B-D) Thrombin generation in normal plasma depleted of CMs (“Thrombin generation assay”) initiated by 10nM (B) α-IIa, (C) β-IIa, or (D) γ-IIa in the presence or absence of 4μM CTI, or the presence of 300 μg/mL O1A6. (E-F) Western blots of (E) PK or (F) FXII (200nM) incubated with vehicle (C) or 25nM α-IIa (α), β-IIa (β), γ-IIa (γ), FXIa (XIa), FXIIa (XIIa), α-kallikrein (Ka), or 0.01nM trypsin (T). Blots were developed with goat polyclonal antibodies against the relevant protein. α-Kallikrein and FXIIa standards (St) are in the first lanes of each blot, with positions of zymogen PK and FXII (Z) and the heavy chain (HC) and light chain (LC) of α-kallikrein and FXIIa shown to the left of each blot. β-kal indicates bands representing the degradation product β-kallikrein. (G-H) Chromogenic substrate assays for activation of (G) PK or (H) FXII (200nM) by 25nM FXIa (●) or vehicle (□) at 37°C. α-Kallikrein or FXIIa generation was assessed by chromogenic substrate assay as described in “Activation of FXII and PK.”
Discussion
Whole genome analyses have provided new insights into the origins of FXI and the contact factors FXII and PK. Although not present in fish, genes for FXII and a predecessor of PK and FXI are found in the frog Xenopus.34,35 A duplication of the hepatocyte growth factor activator gene gave rise to the FXII gene. Interestingly, the apple domains of the FXIIa substrates PK and FXI are homologous to domains on the hepatocyte growth factor activator substrate hepatocyte growth factor. Duplication of the PK/FXI predecessor gene during mammalian evolution produced the PK and FXI genes. The PK/FXI predecessor in the primitive egg-laying mammal the duck-billed platypus is more like PK than FXI.34 Specifically, it appears to consist of a single chain (FXI is a dimer),17 lacks amino acids required for binding FIX,17,34 and has an activation cleavage site more like that of PK than FXI (Table 2). Proline is common at the P2 position in α-IIa substrates (Table 3)9 and is present at P2 in FXI in most species (Table 2). The acquisition by FXI of an activation site suitable for cleavage by thrombin may have been a key step in the transition from PK-like predecessor to coagulation protease.
The active site of α-IIa lies in a cleft flanked by loops that contribute to substrate specificity by restricting access to the active site.1-3 The β-loop (50-insertion loop [Leu49-Asn57]) is a rigid hydrophobic area that interacts with substrates N-terminal to their cleavage sites. The γ-loop (autolysis loop [Leu144-Gly155]) is more mobile and interacts with substrates carboxyl-terminal to the cleavage site. ABE I (Lys21, His66, Arg68, Arg70, Tyr71, Arg73, Lys106, and Lys107) and ABE II (Arg89, Arg98, Arg245, Lys248, and Lys252) further define specificity. Yun et al studied the importance of specific α-IIa amino acids to FXI activation.21 In the presence of dextran sulfate, α-IIa-mediated FXI activation was reduced by alanine substitutions in ABE I (Arg68, Arg70, and Arg73), ABE II (Arg89, Arg98, Arg245, and Lys248), and the β-loop (Trp50). It was concluded that ABE II is required for α-IIa binding to dextran sulfate, whereas ABE I interacts with FXI. Our results with plasma derived and recombinant thrombins in purified protein systems generally agree with those of Yun et al.21 However, in plasma, expression of ABE I activity appears to be detrimental to thrombin-mediated FXI activation. The observation that the ABE I blocking peptide hirugen had a minimal effect on FXI activation by α-IIa in a purified system, and augments FXI-dependent thrombin generation in plasma, argues against a direct binding interaction between FXI and ABE I on α-IIa.
Several proteases are derived from prothrombin during coagulation.9,11 Whereas α-IIa predominates, Mz-IIa, β-IIa, and γ-IIa all form in clotting blood.9,11 The prothrombinase complex initially cleaves prothrombin at the Arg320-Ile321 bond (Figure 1A) forming the short-lived species Mz-IIa.7-10 Using a stable form of Mz-IIa8 in an assay examining fibrin resistance to fibrinolysis, von dem Borne et al were the first to present evidence that this protease activates FXI.22 Consistent with their findings, rMz-IIa has comparable activity to α-IIa in an FXI-dependent thrombin generation assay. rMz-IIa cleaves fibrinogen and PAR-1 less efficiently than α-IIa.10-12,30 Although this suggests that ABE I is not formed, other ABE I–dependent activities, such as FV activation36,37 and thrombomodulin–dependent protein C activation,30 are intact. Anderson and Bock used equilibrium binding methods with hirudin peptides to demonstrate ABE I expression during prothrombin conversion to Mz-IIa.6 ABE I is also present in crystal structures for Mz-IIa(desF1), a protease similar to Mz-IIa but lacking the F1 domain (Figure 1A).31
Conversion of α-IIa to β-IIa involves autoproteolysis within a mobile surface loop that forms part of ABE I (Figure 1B). Trypsin also makes this cleavage, as can FXa or plasmin in plasma. The detrimental effect on ABE I activity is reflected in the reduced capacity of β-IIa to cleave fibrinogen,9,10,38,39 and the 2 orders of magnitude lower affinity for the inhibitor hirudin.39,40 Conversion of β-IIa to γ-IIa involves cleavage in the γ-loop, further impairing fibrinogen cleavage and hirudin binding.9,10,38-40 Dang et al prepared an α-IIa variant lacking the γ-loop (“loopless”) and noted a 240-fold reduction in fibrinogen cleavage compared with α-IIa.41 Based on an analysis of crystal structures, Soslau et al proposed that conversion of α-IIa to β-IIa and then γ-IIa is accompanied by a progressive opening of the active site to take on a more trypsin-like conformation.39 Physiologic roles for β-IIa and γ-IIa have not been established, but several possibilities have been discussed. Although possessing low activity toward fibrinogen and PAR-1, these proteases activate FXIII42 and are inhibited by antithrombin.43 This is not surprising, as the interaction between α-IIa and these proteins does not require ABE I (Table 1). β-IIa and γ-IIa at nanomolar concentrations are also activators of PAR-4 on platelets.39
The importance of ABE I is obvious in thrombin-triggered clotting assays run in the absence of calcium. Here, the added thrombin must covert fibrinogen to fibrin. β-IIa has only 3% of the activity of α-IIa, and γ-IIa does not have measurable activity. Restoring calcium allows thrombin generation from plasma prothrombin to occur, facilitating fibrin formation in plasma stimulated with rMz-IIa, and β-IIa and γ-IIa, but not with α-IIa. This probably reflects the substantially greater efficiency with which α-IIa generates fibrin. The weaker activities of rMz-IIa, β-IIa and γ-IIa toward fibrinogen require α-IIa generated from endogenous prothrombin for optimal performance in the clotting assay. Semenova and Strukova observed that β-IIa and γ-IIa induced fibrin formation required calcium, consistent with our results.44 For rMz-IIa, FXI is not involved in initial clot formation. Although the proteolytic target through which rMz-IIa enhances endogenous α-IIa production is not certain, FV would be a strong candidate. For β-IIa and γ-IIa, optimal fibrin formation depends on FXI. Indeed, with γ-IIa, FXI is required for fibrin formation. Taken as a whole, the clotting data support the premise that β-IIa and γ-IIa are activators of FXI in plasma, despite the loss of expression of ABE I activity.
Fibrin formation occurs relatively early in the coagulation process and requires relatively small amounts of α-IIa. However, the bulk of thrombin formation, as reflected in the TGA, occurs after the fibrin clot has formed. It is here that the impressive effects of β-IIa and γ-IIa thrombin on FXI-dependent thrombin generation are clearly demonstrated. β-IIa and γ-IIa were consistently approximately 1.5- to 2-fold better initiators of FXI-dependent thrombin generation than α-IIa (or rMz-IIa) in the presence or absence of FXII. These data, in conjunction with those for α-IIa-Glu68 and α-IIa-Glu70, and with the ABE I-blocking peptide hirugen, raise the possibility that weak ABE I expression for β-IIa or γ-IIa reduces the capacity of fibrinogen and other substrates that interact with ABE I to compete with FXI for access to the protease. Indeed, fibrinogen was shown to inhibit α-IIa-mediated FXI activation in a purified system.45 Fibrin is designated antithrombin I because of its capacity to reduce α-IIa activity through nonsubstrate interactions.46 A low-affinity interaction between ABE I and the fibrin E domain facilitates a high-affinity interaction between ABE II and fibrin γ′ chains, down-regulating α-IIa activity. Loss of antithrombin I activity has been implicated in thrombotic episodes in patients with afibrinogenemia47,48 and some dysfibrinogenemias.49 β-IIa and γ-IIa are probably less susceptible to antithrombin I activity, partly explaining their potency in the thrombin generation assay.
TGA assays typically include a FXIIa inhibitor to prevent FXIIa-induced spontaneous thrombin generation. Our observation that plasma CMs may be responsible for this process, at least in part, has clinical implications, in addition to allowing us to study FXI-dependent thrombin generation in uninhibited normal plasma. There are data suggesting that triglyceride-rich lipoproteins (CMs and very low density lipoproteins) support contact activation, possibly through charged head-groups in their phospholipid outer layers.50,51 This may explain an association noted between some types of hyperlipidemia and elevated plasma FXIIa levels, and raises the possibility that lipid-induced contact activation could contribute to thrombin generation and risk for thrombotic disease.52 CM-depleted plasma did not demonstrate spontaneous thrombin generation in our TGA, even with prolonged incubation, indicating endogenous basal levels of active FXIIa are low. However, when FXI-dependent thrombin generation was induced with α-, β-, or γ-IIa, FXIIa still contributed significantly to the process. Whereas α-, β-, or γ-IIa do not appear to convert FXII to FXIIa, FXIa does catalyze this reaction. FXIa may, therefore, target FXII as well as FIX in plasma, with FXIIa impacting thrombin generation through FXI activation.
FXIa is a homolog of the potent FXII activator α-kallikrein. Previously, Griffin reported that FXIa activates factor XII, although with an order of magnitude lower efficiency than α-kallikrein.53 The mechanism behind FXII activation in vivo has long been a topic of discussion. During contact activation-induced coagulation initiated by a surface, reciprocal activation of FXII and PK drives FXIIa formation and subsequent generation of FXIa.18,53 Recent studies have implicated polyphosphates released from platelet granules as a physiologic surface for contact activation54 ; however, it is not clear whether FXII activation on any surface is triggered by a conformational change conferring proteolytic activity to zymogen FXII, or by traces of FXIIa or other proteases (eg, α-kallikrein) present in blood or released from cells. Our results raise the possibility that generation of FXIa by thrombin during a hemostatic response to tissue injury could contribute to FXII activation, providing a link between tissue factor-initiated blood coagulation and the contact system.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Drs Paul Bock and Enrico Di Cera for helpful discussions on thrombin structure and thrombin generation assays.
This work was supported by the National Heart, Lung and Blood Institute (HL81326 and HL58837, D.G.; and HL080018, I.M.V.).
National Institutes of Health
Authorship
Contribution: A.M. conducted plasma clotting studies, FXI activation studies by SDS-PAGE, thrombin generation studies, analysis of FXI and PK activation, and manuscript preparation; S.S. prepared and characterized recombinant Mz-IIa and was involved in its analysis; M.-f.S. prepared recombinant FXI proteins and conducted FXI activation studies by SDS-PAGE; J.P.S. prepared recombinant α-thrombin variants and helped design studies; V.S. assisted in designing and supervising studies; I.M.V. prepared and characterized recombinant Mz-IIa, assisted in study design and result interpretation, and assisted with manuscript preparation; and D.G. supervised the project and prepared the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David Gailani, Division of Hematology/Oncology, Vanderbilt University, 777 Preston Research Bldg, 2220 Pierce Ave, Nashville, TN 37232-6305; e-mail: dave.gailani@vanderbilt.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal