

Abstract
Mathematical models of chronic myeloid leukemia (CML) cell population dynamics are being developed to improve CML understanding and treatment. We review such models in light of relevant findings from radiobiology, emphasizing 3 points. First, the CML models almost all assert that the latency time, from CML initiation to diagnosis, is at most ∼ 10 years. Meanwhile, current radiobiologic estimates, based on Japanese atomic bomb survivor data, indicate a substantially higher maximum, suggesting longer-term relapses and extra resistance mutations. Second, different CML models assume different numbers, between 400 and 106, of normal HSCs. Radiobiologic estimates favor values > 106 for the number of normal cells (often assumed to be the HSCs) that are at risk for a CML-initiating BCR-ABL translocation. Moreover, there is some evidence for an HSC dead-band hypothesis, consistent with HSC numbers being very different across different healthy adults. Third, radiobiologists have found that sporadic (background, age-driven) chromosome translocation incidence increases with age during adulthood. BCR-ABL translocation incidence increasing with age would provide a hitherto underanalyzed contribution to observed background adult-onset CML incidence acceleration with age, and would cast some doubt on stage-number inferences from multistage carcinogenesis models in general.
Introduction
Chronic myeloid leukemia (CML) is characterized by Ph+ cells, that is, cells having a Philadelphia (BCR-ABL) chromosome translocation.1,2 Treatment with the tyrosine kinase inhibitor (TKI) imatinib mesylate (“imatinib”), which suppresses bcr-abl oncoprotein action,3 improves patient prognosis dramatically.4 However, in some cases this treatment fails, a problem mitigated but not fully solved by the use of more recently developed TKI.5,6 Moreover, many patients may need to continue TKI treatment indefinitely to avoid relapse.7
CML is one of the best understood cancers; it has a simpler etiology than most cancers8 and its time course is comparatively easy to monitor in the clinic.9,10 Consequently, despite being much less prevalent than major solid tumors, CML has often been regarded as a kind of “model organism” for quantitative modeling of human carcinogenesis.11,12
CML cell population dynamics
In this review, we emphasize how radiobiologic studies impact CML models grounded in understanding underlying cell population dynamics. These models track CML time evolution by differential equations and/or stochastic formalisms. Such biologically based quantitative models are more ambitious, more comprehensive, and as yet less definitive than models often used in statistical analyses, which emphasize correlations analyzed by adjusting parameters in functions chosen mainly for mathematical convenience.
After work by Rubinow and Lebowitz13 on hematopoiesis, biologically based, mathematical CML models were pioneered by Clarkson and coworkers.14 Many additional models have been suggested in the last decade. Recent articles include work by Michor and coworkers,15,16 Dingli and coworkers,17 Roeder and coworkers,18 Levy and coworkers,19,20 and many others (reviewed in Whichard et al9 and Roeder and d'Inverno,21 and in supplemental Section 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Importantly, such models can generate useful new insights by quantitatively interrelating datasets that seem offhand to have little connection with each other, let alone to have systematic numerical correlations. There are several examples, including (1) the successful application of data on hematopoiesis in healthy individuals to predictions on the time course of the molecular response in TKI-treated CML patients15 ; (2) the application of parameters calibrated by data on the time course of the molecular response in imatinib-treated patients to data on the time course of blast crisis onset in preimatinib-era patients22 ; and (3) applying data on ionizing-radiation–induced CML to modeling background (ie, sporadic) CML, as documented in this review. Without biologically based modeling, it is often hard to see such connections, much less quantify them.
Parameter calibrations for comprehensive, biologically based, quantitative CML models require consideration of many different datasets and are typically far more difficult than devising and manipulating the mathematical/computational formalism. The underlying idea is to calibrate a multiscale model as a whole, using a broad range of clinical, epidemiologic, and (when necessary) auxiliary laboratory data. If credible parameter calibration can be achieved, it becomes comparatively easy to make critical comparisons with other models, test a model's extrapolations using additional data, make testable predictions for almost any relevant kind of future datasets, and hopefully even gain a deeper understanding of carcinogenesis in general.
Radiobiology
Radiobiology is highly suited for dissecting mechanistic CML studies because ionizing radiation, which is known to induce CML23 that appears indistinguishable from background CML,24 is an unusually well-understood carcinogen, especially at small time and length scales. The following radiation properties have been characterized and mathematically modeled with relatively high precision: radiation track structure (reviewed in Friedland25 ); micro- and nanodosimetry (reviewed in Grosswendt et al26 and Hei et al27 ); inter- and intratissue dose distributions, such as human dose-volume histograms28 ; radiation action at submillisecond times (reviewed in Wardman29 ); subsequent DNA damage-repair-misrepair mechanisms30 ; DNA damage-processing outcomes (eg, chromosomal aberrations,31 which are especially important here because of their relevance to BCR-ABL translocations,32 gene mutations,33 and transcriptome changes34 ); and many additional relevant end points.35 Overall, radiation perturbations in humans are frequently more informative than chemical perturbations, because of: (1) better known dose localization, both spatially and temporally; (2) thorough knowledge of particle-track physics; and (3) the ability to change not only the dose but also the ionizing particle type and/or energy,36 with resulting changes in response giving extra information. Importantly, the specific initiation time for a radiation-induced cancer is often well-defined.
The “gold standard” for analyses of ionizing radiation-induced cancers is the continuing lifespan study (LSS; reviewed in Douple et al37 and Ozasa et al38 ) of the Japanese survivors of the atomic bombs dropped in 1945. The population affected was large and more nearly representative of normal human demographics than in other studies of radiation carcinogenesis. Much of this dataset is publicly available. The many different articles on the LSS focus primarily on radiation dose-response, but also contain information, highly pertinent here, on how CML incidence and mortality depend on age at exposure and time since exposure.
Approach
This review describes how radiobiology relates to biologically based, quantitative CML models. CML modeling emphasizes clinical issues such as response to TKI treatment, while most of the relevant radiobiology data are epidemiologic and concern mainly the time before CML diagnosis. Issues that are clinical without being directly related to radiobiology are therefore reviewed separately, as background, in supplemental Section 1. The review itself will focus instead on how radiobiology data can be used to inform the development of quantitative CML models and assist in their parameter calibration, thereby helping lay the groundwork for clinical analyses.
Throughout, as is appropriate in a review, the discussion emphasizes CML models and radiobiology data in their published versions, without considering possible improvements. However, sometimes elementary calculations based on publicly available datasets elucidate the interrelation between radiobiology and CML. In such cases, we used the calculations, avoiding additional assumptions as much as possible. Supplemental Section 3 gives links to R scripts implementing the calculations.
Quantitative CML models
Biomathematic implementations
Recent biomathematic CML models track cell- and tissue-level effects over several years, emphasizing changes of cell numbers in various hematopoietic subpopulations, leukemic and normal (Figure 1A).

Cell population dynamic CML models. (A) As an example, one recent model39 considers 5 leukemic cell subpopulations, as shown. For normal (ie, Ph−) cells and for TKI-resistant mutants, similar diagrams are postulated. Arrows indicate possible ways cells can move to a different subpopulation by differentiation or change of cycling status. Parameter calibration requires estimating rate parameters for proliferation, differentiation, and cell death, some of which are affected by TKI treatment such as imatinib dosing. Other current quantitative CML models differ in detail. Some consider many more subpopulations; some do not distinguish between quiescent and cycling LSCs, etc. But most of the models similarly emphasize cell population dynamics and TKI treatment. Some models also consider immune system interactions with CML (supplemental Section 1.6) or some molecular-level events, including CML initiation and/or alterations that may drive CML progression. (B) A “typical” timeline for CML. In this review, CML “initiation“ will refer to the origination of a Ph+ LSC clone sufficiently large that the probability of accidental extinction has become negligible. Current models typically assume that once such a clone has become established, clinically diagnosable CML will result after some, in general patient-dependent, latency time. Parameters relevant to the latency time are often especially problematic since little direct information is available: in humans, CML initiation and early evolution are usually cryptic, although some information about cell population dynamics toward the end of the latency time can be gathered from patients who are diagnosed at atypically early phases of CML.12
Cell population dynamic CML models. (A) As an example, one recent model39 considers 5 leukemic cell subpopulations, as shown. For normal (ie, Ph−) cells and for TKI-resistant mutants, similar diagrams are postulated. Arrows indicate possible ways cells can move to a different subpopulation by differentiation or change of cycling status. Parameter calibration requires estimating rate parameters for proliferation, differentiation, and cell death, some of which are affected by TKI treatment such as imatinib dosing. Other current quantitative CML models differ in detail. Some consider many more subpopulations; some do not distinguish between quiescent and cycling LSCs, etc. But most of the models similarly emphasize cell population dynamics and TKI treatment. Some models also consider immune system interactions with CML (supplemental Section 1.6) or some molecular-level events, including CML initiation and/or alterations that may drive CML progression. (B) A “typical” timeline for CML. In this review, CML “initiation“ will refer to the origination of a Ph+ LSC clone sufficiently large that the probability of accidental extinction has become negligible. Current models typically assume that once such a clone has become established, clinically diagnosable CML will result after some, in general patient-dependent, latency time. Parameters relevant to the latency time are often especially problematic since little direct information is available: in humans, CML initiation and early evolution are usually cryptic, although some information about cell population dynamics toward the end of the latency time can be gathered from patients who are diagnosed at atypically early phases of CML.12
Many of the CML models are stochastic, that is, probabilistic (reviewed in Whichard et al9 ). Especially in situations where the number of cells in some subpopulation is small, interpatient response differences may require stochastic calculations, rather than just deterministic calculations which concern averages (Figure 2). For example, if a person harbors only a few Ph+ leukemic stem cells (LSCs), an LSC-free state can result even if on average LSCs have a growth advantage over HSCs, because Ph+ clones can become extinct by “chance” (eg, Figure 2 patient 7). Estimating extinction probabilities requires stochastic models. However, if all cell subpopulation average numbers are always ≫ 1, differences between deterministic and stochastic models are often minor.

TKI discontinuation possibilities illustrate deterministic versus stochastic modeling differences. Suppose that: (1) CML LSCs have a growth advantage over HSCs in the absence of treatment, but a growth disadvantage under TKI treatment; and (2) treatment is discontinued when the average number of LSCs is just one. What will happen? On a deterministic model, all patients will eventually relapse, as the LSCs repopulate (here schematically indicated as exponential growth). On a more realistic stochastic model, the results are quite different, even if the average number of LSCs at each time equals the deterministic estimate at that time. Here, patients 1 and 2 were “lucky” enough to be fully LSC-free when treatment was discontinued; afterward, they remain disease-free. “Luck” refers to factors not systematically foreseeable with current techniques. Patient 7 was even luckier: both LSC clones died out accidentally, one after the other, despite their growth advantage. On the other hand, patient 8 is substantially worse off than the deterministic model would predict, while patients 5 and 6 have also been rather unlucky. Whether any of the 5 LSC-bearing patients will later suffer a clinical relapse without treatment is in part a matter of luck, but patient 8 has a comparatively large chance of relapse.
TKI discontinuation possibilities illustrate deterministic versus stochastic modeling differences. Suppose that: (1) CML LSCs have a growth advantage over HSCs in the absence of treatment, but a growth disadvantage under TKI treatment; and (2) treatment is discontinued when the average number of LSCs is just one. What will happen? On a deterministic model, all patients will eventually relapse, as the LSCs repopulate (here schematically indicated as exponential growth). On a more realistic stochastic model, the results are quite different, even if the average number of LSCs at each time equals the deterministic estimate at that time. Here, patients 1 and 2 were “lucky” enough to be fully LSC-free when treatment was discontinued; afterward, they remain disease-free. “Luck” refers to factors not systematically foreseeable with current techniques. Patient 7 was even luckier: both LSC clones died out accidentally, one after the other, despite their growth advantage. On the other hand, patient 8 is substantially worse off than the deterministic model would predict, while patients 5 and 6 have also been rather unlucky. Whether any of the 5 LSC-bearing patients will later suffer a clinical relapse without treatment is in part a matter of luck, but patient 8 has a comparatively large chance of relapse.
Stochastic models are often implemented by Monte Carlo simulations,40 which are very intuitive and allow extraordinary flexibility in model assumptions. When possible, the simulations are supplemented with equations from stochastic process theory,41 which can give global insights into dependence of model predictions on adjustable parameters, check the simulations, and speed them up.
Similarities and differences among models
The models usually assume that CML LSCs are HSCs that have acquired a BCR-ABL translocation. They emphasize predictions on cell subpopulation changes after CML diagnosis, for example, the following: hematopoietic, cytogenetic, and molecular responses to TKI treatments; presentation of mutant clones resistant to TKI treatment; responses to treatment discontinuation or alteration; immune system responses to CML; CML responses to cocktails of different drugs; and long-term CML management.
Differences among the models include the following:
whether TKI are considered to act on LSCs as opposed to acting only on more differentiated Ph+ cells (reviewed in Roeder and d'Inverno21 );
estimates, in some cases differing by multiplicative factors > 103, of the number of HSCs17,39 ;
the latency time (Figure 1B) postulated or predicted, as discussed in the next section;
whether spatial inhomogeneities, considered to be key factors in solid tumors,44 and probably important within the BM for CML,45 are modeled; and
whether interactions between Ph+ cells and the immune system are taken into account.20,46
Reconciling such differences among the models credibly calls for comparisons using common datasets, which can only be done after each model is credibly calibrated.
Data considered
The data reviewed here are primarily human in vivo results, which should, whenever possible, be emphasized in modeling clinically relevant cell population dynamics. In the next 3 sections, 3 main types of data will be considered, as follows:
LSS data on the timing of radiation-induced CML in the Japanese atomic bomb survivors (reviewed in UNSCEAR47 and Preston et al48 ), which provide the most nearly direct information on the duration of CML latency in humans.
SEER cancer database data49 on age dependence of background CML incidence; and, for comparison, data on age dependence of background chromosome translocations developed to assist radiation biodosimetry (reviewed in Edwards et al50 and Sigurdson et al51 ).
Corresponding results on radiation-induced translocations (reviewed in Tucker52 ), relevant to estimates of the number of normal “LSC-predecessor target” cells at risk for a CML-initiating BCR-ABL translocation.
Many datasets on radiogenic cancers group together all leukemias except chronic lymphocytic leukemia (which is considered nonradiogenic). CML normally makes up less than one-third of the grouped total and is known to behave differently from the other major subtypes. Therefore, data grouped in this way is not considered in this CML-specific review. Apart from this limitation, we strove to consider all relevant radiobiologic literature, and to reference representative examples. In particular, both CML mortality and incidence data, which are to some extent complementary,53 are included. The relevant mortality data are from the preimatinib era when, barring BM transplantations, mortality almost always followed diagnosis in < 10 years,54 so one would expect considerable concordance. However, as will be seen, there are some discrepancies.
CML latency
Radiobiology48,55-57 and comparisons of humans with other mammals58,59 have been used to estimate the CML latency time T (Figure 1B), an important parameter in mathematical CML models. The radiobiology estimates (Figure 3) identified latency time with the time interval between an acute ionizing radiation exposure and CML diagnosis.

CML latency time T. T is here considered as a random variable, that is, a quantity described by a probability density because it may fluctuate from case to case in a way not (yet) fully predictable. In light of an early report56 on the Japanese atomic bomb survivors, clinically oriented quantitative CML models predict or assume a density that is narrow (has small SD), for example, the red curve. However, more recent LSS results indicate broader curves, having substantially larger SD and extended upper tails, for example, the blue curve. This curve is based on male CML incidence in the LSS database up to 1987, for doses between 0.01 and 4 Sievert (Sv; 1 Sv is defined as that dose which has the same biologic effect as 1 Gy of hard x-rays, where 1 Gy = 1 Joule/kg), with ∼ 15 of 22 total cases believed to be radiation-induced.48 As discussed in the paragraph before this figure and the paragraphs below it, other current radiobiologic estimates are even broader than the blue curve and their right tails extend out even farther. Details on latency time estimates in many different CML models and on the methods used to calculate the curves shown are in supplemental Section 2.2.
CML latency time T. T is here considered as a random variable, that is, a quantity described by a probability density because it may fluctuate from case to case in a way not (yet) fully predictable. In light of an early report56 on the Japanese atomic bomb survivors, clinically oriented quantitative CML models predict or assume a density that is narrow (has small SD), for example, the red curve. However, more recent LSS results indicate broader curves, having substantially larger SD and extended upper tails, for example, the blue curve. This curve is based on male CML incidence in the LSS database up to 1987, for doses between 0.01 and 4 Sievert (Sv; 1 Sv is defined as that dose which has the same biologic effect as 1 Gy of hard x-rays, where 1 Gy = 1 Joule/kg), with ∼ 15 of 22 total cases believed to be radiation-induced.48 As discussed in the paragraph before this figure and the paragraphs below it, other current radiobiologic estimates are even broader than the blue curve and their right tails extend out even farther. Details on latency time estimates in many different CML models and on the methods used to calculate the curves shown are in supplemental Section 2.2.
Radiobiologic data
A 1981 article on 42 cases of CML diagnosed during 1950-1978 in a population consisting mainly of Japanese atomic bomb survivors, has been influential in estimating CML latency periods.56 The article reported that CML incidence peaked < 10 years after the 1945 radiation exposure. However, the data were consistent with some excess risk up to 30 years after exposure, and it did not tabulate results for the first 5 years after exposure. Subsequent studies of CML incidence in the Japanese LSS population extended the time period in both directions, stratified by sex and city, used improved dosimetry, and explicitly estimated the percentage of cases that were radiation-induced. These later studies point to a much broader distribution of latency times. The blue curve in Figure 3 shows one current radiobiologic estimate, narrower than other current radiobiologic estimates, but substantially broader than the estimates used in cell population dynamic CML models.
Analyses38,55 of LSS CML mortality data through the year 2000 (58 deaths, with ∼ 25 attributed to radiation) even conclude that CML excess absolute risk (EAR) is independent of time since exposure (and is sex independent). These data are not yet publicly available, but prima facie the conclusions seem to indicate that some CML cases initiated by the atomic bombs might not present for a very long time, ∼ 50 years.
Using a simple statistical approach, the publicly available LSS data can be viewed in a way that sheds some light on the differences between the radiobiologic incidence and mortality studies. The approach fits CML incidence as the sum of a background incidence that increases exponentially with age (as justified in the next section), plus, as is appropriate for CML32,48,55 (though not for leukemias as a group), an excess response linear in radiation dose D:
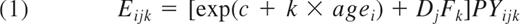
Here: PY = person-years; Eijk is the expected number of CML cases for the ijkth data cell (ith attained-age group, jth dose group, and kth time-since-exposure group); Fk are linear dose-response slope estimates for different times since exposure (7, 13, 21, 31, and 40 years; 10 groups in the data, not all of equal bin widths, were paired in sequence to form these 5 groups); and Dj are independently reconstructed doses given in the dataset. This model was fitted separately for males and females. The resulting Fk (ie, the EAR estimates) are shown in Figure 4. In agreement with Preston et al,48 these EAR estimates suggest that, compared with males, female CML incidence is more nearly consistent with an EAR that is independent of time since exposure, though even for females the EAR decreased markedly after 40 years. Thus, the discrepancy between radiogenic incidence and mortality estimates remains; speculating, plots analogous to Figure 4, but based on CML mortality, may reveal an EAR rise in the 13 years of additional follow-up (1987-2000), perhaps allowing a rough approximation as an overall horizontal line. In any case, current LSS incidence and mortality data are both clearly inconsistent with any waiting time curve narrower than the blue curve in Figure 3.

Atomic bomb survivor waiting times. The EAR parameters Fk of Equation (1), fitted separately to male and female atomic bomb survivor data, are shown. The results indicate that radiation-induced CML incidence decreases with time. Male latencies are < 20 years and female latencies are bimodal with one mode at ∼ 5 years (similar to males) and the other at ∼ 32 years. The Wald 95% confidence interval of the female Fk parameter shown at ∼ 32 years does not include the corresponding male point.
Atomic bomb survivor waiting times. The EAR parameters Fk of Equation (1), fitted separately to male and female atomic bomb survivor data, are shown. The results indicate that radiation-induced CML incidence decreases with time. Male latencies are < 20 years and female latencies are bimodal with one mode at ∼ 5 years (similar to males) and the other at ∼ 32 years. The Wald 95% confidence interval of the female Fk parameter shown at ∼ 32 years does not include the corresponding male point.
Many radiobiologic studies of other populations (reviewed in UNSCEAR57 ) concern radiation exposures which are environmental, occupational,60 accidental, or related to medical treatment/diagnostics.24 No clear inconsistencies between the LSS and these other studies have been found.61,62 A recent study on > 350 000 women who underwent radiation therapy for breast cancer estimated that CML incidence was elevated as late as 25 years after breast cancer radiotherapy.63 In general, however, inadequate statistical power and/or the common practice of grouping CML with other leukemias prevent non-LSS studies from furnishing tight quantitative information on the probability distributions of CML latency times.57
Thus, radiobiologic analyses show that CML incidence peaks within 10 years after radiation, but several decades, and perhaps a half-century, can separate CML initiation and diagnosis (Figures 1B, 3, 4). Why is the variation in latency times so big? One suggestion has been that in addition to initiating CML, radiation might speed up later carcinogenesis steps,55 for example, by acting on previously initiated CML LSCs or by killing some normal HSCs which could then get replaced, preferentially because of their growth advantage, by previously initiated LSCs (compare Nakamura,64 Laukkanen et al,65 and Little et al66 ). For example, suppose, hypothetically, that the mean of the true latency time, from initiation to diagnosis, is ∼ 30 years, and that ∼ 25 years after initiation there is a promotion bottleneck which radiation can help overcome. Then, an acute dose would result in an EAR curve which has a hump, corresponding to radiation initiation, at ∼ 30 years. But there would also be another hump at ∼ 5 years, corresponding to radiation promotion of LSCs which had been initiated, by other mechanisms, ∼ 25 years before the dose. In this case, the EAR curve would somewhat resemble the female EAR curve in Figure 4, and using it would: (1) underestimate the true mean latency time; (2) overestimate the SD; but (3) give an accurate portrayal of the rightward tail that, as we argue below, may well be clinically relevant.
Importance for CML models
In contrast to the radiobiologic results discussed in connection with Figure 3, CML models used in analyzing TKI treatments either assume or predict quite narrow probability densities for T (see supplemental Section 2.2 for details). Deterministic CML models usually even have, in effect, infinitely narrow probability densities (SD = 0). The only CML model we found that has a large rightward tail in the latency time distribution was one designed to analyze background CML as a function of age,67 which uses a waiting time approach considerably different from clinically oriented CML models.
For some important applications of the CML models, such as their mechanistic explanations of the decline in BCR-ABL signal during the first year of first-line TKI treatment, underestimation of the latency time distribution rightward tail makes little difference. Latency times do, however, enter, directly or indirectly, into many clinically relevant predictions,12 especially for the possibility of relapse after discontinuation or interruption of kinase inhibitor treatment subsequent to complete molecular remission.20 Most quantitative CML models imply that, if T has a large mean and/or SD, relapses can still occur long after the treatment has stopped. And some models further predict that a large latency time SD is associated with a higher probability of developing TKI resistance (reviewed in Katouli and Komarova68 ; the mathematical rationale for this association is outlined in supplemental Section 1.4). Thus, improving latency time estimates is important. The ongoing STIM (Stop Imatinib Clinical Trial) trials,7 other imatinib discontinuation studies,69 and the ongoing LSS study37 will continue to give additional quantitative information on these key points, so that CML modeling and radiobiology will help inform each other regarding latency times and their variance–a rather remarkable example of how biologically based quantitative modeling can interrelate prima facie unrelated datasets.
Comparing age dependence of CML and of chromosome translocations
The background incidence of adult-onset CML accelerates exponentially with age (Figure 5A). A comprehensive, biologically based CML model should aim to explain the observed acceleration.67 Such explanations have been attempted for many cancers using multistage models (reviewed in Little et al66 and Fakir et al70 ). Quantitative models that analyze CML treatment have not yet incorporated analyses of CML incidence with age. This section discusses results, relevant to any such unification, on how background in vivo chromosome translocation levels increase with age.

Age dependence of background CML incidence and translocation prevalence. (A) Data49 are for background CML during 1973-2008 and for age at diagnosis > 20 years. Data are shown here in a semilogarithmic plot with straight line fits y = Aek*age; female and male k are almost equal. (B) One would expect background chromosomal translocations to correlate with CML incidence because CML is caused by a specific translocation. The data for this plot of cumulative translocation clones in peripheral blood lymphocytes of healthy individuals were obtained (using plotDigitizer) from Figure 4 of Sigurdson et al.51 Evidence for exponential behavior is not as convincing as in panel A: a significant improvement in fit (P = .015, F or t test) is obtained with an additional a2 term, consistent with slight curvature in the data that is visible by inspection. (C) A log-log plot of the same data as in panel B, with the fitted straight line given by y ∝ (age)1.5. As discussed in “Conclusions,” the translocation results seem inconsistent with a standard assumption in the multistage models usually used to explain age-driven increases in tumor incidence.
Age dependence of background CML incidence and translocation prevalence. (A) Data49 are for background CML during 1973-2008 and for age at diagnosis > 20 years. Data are shown here in a semilogarithmic plot with straight line fits y = Aek*age; female and male k are almost equal. (B) One would expect background chromosomal translocations to correlate with CML incidence because CML is caused by a specific translocation. The data for this plot of cumulative translocation clones in peripheral blood lymphocytes of healthy individuals were obtained (using plotDigitizer) from Figure 4 of Sigurdson et al.51 Evidence for exponential behavior is not as convincing as in panel A: a significant improvement in fit (P = .015, F or t test) is obtained with an additional a2 term, consistent with slight curvature in the data that is visible by inspection. (C) A log-log plot of the same data as in panel B, with the fitted straight line given by y ∝ (age)1.5. As discussed in “Conclusions,” the translocation results seem inconsistent with a standard assumption in the multistage models usually used to explain age-driven increases in tumor incidence.
The translocation data presented in Figure 5 were generated as controls for radiation biodosimetry (reviewed in Edwards et al,50 Sigurdson et al,51 and Tucker52 ) and do not single out BCR-ABL. In these data, accumulated numbers of clones with translocations are counted at various ages; a clone with a specific translocation is counted only once no matter how many cells the clone has. This procedure gives translocation clone prevalence P(a) rather than incidence I(a), where a is attained age. However, in vivo translocation clones tend to decay away gradually71 (reviewed in Tucker52 ), perhaps because of a growth disadvantage on average (contrasting with the growth advantage of Ph+ clones in the absence of TKI treatment). The decay implies that prevalence and incidence have similar behavior.72 For example, for exponential decay (ie, first-order linear kinetics) with rate r > 0, the derivative of prevalence is

Equation 2 implies that if P(a) ∝ exp[ka] with k > 0 then I(a) = rP + P′ ∝ exp[ka] also; the same argument holds for the most rapidly increasing term in I if P(a) ∝ (a)k. Thus, we can, and shall, compare observed CML incidence with observed translocation prevalence to estimate whether BCR-ABL clone incidence, assumed in this argument to track overall translocation clone incidence, could have an important influence on CML incidence acceleration.
Translocation clone prevalence during adulthood increases faster than linearly and perhaps exponentially (Figure 5B). Standard least squares regression of an exponential to recent pooled data51 for ages > 20 years yields a value of k ∼ 0.031 per year, comparable with an earlier estimate73 of 0.04 per year using earlier data.74 Alternatively, assuming the increases shown in Figure 5 are proportional to a power of age, rather than being exponential, leads to a somewhat worse fit for CML incidence (supplemental Figure 4) but a better one for translocation clone prevalence (Figure 5C). For CML incidence, the power is ∼ 2.5 (our estimate here) to ∼ 2.86 (estimated in Michor et al67 ); for translocation clone prevalence, the estimated power is ∼ 1.5 (our estimate here) to almost 2 (estimated in Lucas75 ). In any case, the growth parameters for background CML incidence and for background translocation incidence (as inferred from translocation prevalence) are substantially but not drastically different, a result whose possible implications are far-reaching (see “Conclusions”).
Estimating LSC-predecessor cell numbers for CML
For CML, how many normal cells are “LSC-predecessor” cells (sometimes called “target” cells or “CML initiating” cells), that is, normal cells at risk of a BCR-ABL translocation that can initiate CML by starting an LSC clone? CML models often answer by assuming LSC-predecessor cells are HSCs, whose number can be estimated.76 Chromosome translocation data offer an alternative approach to this question, as we now discuss.
Radiobiologic and background data
To date, radiation-induced CML incidence has not been found to have significant dependence on age at exposure,32,48,55 suggesting approximate age independence during adulthood for the number of LSC-predecessor cells (and perhaps even during childhood for the “effective number,“ ie, the actual number corrected for extra postinitiation LSC growth at young ages77 ). This pattern is shown graphically in Figure 6 by plotting CML incidence versus age at exposure for low-, medium- and high-dose atomic bomb survivors. The high-dose group contains 12 CML cases where 0.8 background cases are expected, so these cases are almost all radiation-induced; that is, they correspond to the DjFk term in equation 1. The approximate constancy seen for this high-dose group across age at exposure is consistent with earlier estimates32,48,55 of age-at-exposure independence.

CML incidence versus age at radiation exposure for different dose groups. Japanese atomic bomb survivors were partitioned into 3 dose groups, low (D < 0.02 Sv), medium (0.02 Sv < D < 1 Sv), and high (D > 1 Sv), and 3 age-at-exposure groups age < 20 years, 20 < age < 40 years, and age > 40 years. Person-year weighted averages of age at exposures are plotted on the x-axis; CML cases diagnosed between 1950 and 1987 divided by corresponding person-years are shown on the y-axis. In the high-dose group, incidences are 1.5, 3.2, and 1.8 per 104 person-years at average age at exposures of 10.9, 29.0, and 47.4 years. In the low- and medium-dose groups, small values for childhood exposures are mainly due to the fact that people in this age group were just reaching ages of high incidence in 1987, that is, approximate independence of age at exposure is likely to hold even better when post-1987 data are added..
CML incidence versus age at radiation exposure for different dose groups. Japanese atomic bomb survivors were partitioned into 3 dose groups, low (D < 0.02 Sv), medium (0.02 Sv < D < 1 Sv), and high (D > 1 Sv), and 3 age-at-exposure groups age < 20 years, 20 < age < 40 years, and age > 40 years. Person-year weighted averages of age at exposures are plotted on the x-axis; CML cases diagnosed between 1950 and 1987 divided by corresponding person-years are shown on the y-axis. In the high-dose group, incidences are 1.5, 3.2, and 1.8 per 104 person-years at average age at exposures of 10.9, 29.0, and 47.4 years. In the low- and medium-dose groups, small values for childhood exposures are mainly due to the fact that people in this age group were just reaching ages of high incidence in 1987, that is, approximate independence of age at exposure is likely to hold even better when post-1987 data are added..
Assuming then that LSC-predecessor cell numbers are approximately constant across adult age groups, estimates of this constant can be derived for age-induced, and independently for radiation-induced, incidence using estimates for total translocations and CML. For the age-induced case, relevant data on translocations is fairly firm51 (see Figure 5B), and CML data are very firm.49 In the radiation-induced case, firm results on translocations are available both theoretically and experimentally50-52,78 ; the relevant data on CML48 has been discussed in connection with Figure 5.
In addition to these results, one needs only the conditional probability P(BA|trans) that a cell acquires a BCR-ABL translocation given that it acquires a translocation. P(BA|trans) can be estimated to be twice the product of the relevant BCR and ABL intron sizes divided by the genome size squared.79,80 The appropriate comparisons of radiation-induced CML and translocation probabilities32,80 as well as of age-induced CML and translocation probabilities73 then yield similar estimates of the number of LSC-predecessor cells, namely ∼ 1 × 108. Thus, there are 2 situations (age-induced vs radiation-induced damage), and for each situation 2 end points (CML and translocations); therefore, there are 2 independent estimates of LSC-predecessor cell numbers. These 2 agree approximately with each other.
This P(BA|trans) is, however, too simplistic, as there is interphase fluorescence in situ hybridization BCR-to-ABL distance data for nonleukemic cells suggesting tethering between chromosomes 9 and 22 that creates unusually close distances between BCR and ABL.81,82 One implication of this spatial proximity (Figure 7) is an increased estimate for the probability of BCR-ABL translocations per cell, with a corresponding decrease of LSC-predecessor cell number estimates to values in the range of 5 × 106 to 1 × 107 (see Radivoyevitch et al32 ). These values are consistent with ∼ 1012 nucleated marrow cells83 and one long-term initiating cell per 105 nucleated marrow cells.84 They are, however, inconsistent with estimates of ∼ 11 000 HSCs,59,76 less inconsistent with values 20-fold higher obtained using the NOD/SCID assay,85 and perhaps borderline consistent if female NOD/SCID/γ recipient mice are used.86

Proximity effects. (A) Schematically shows 1 chromosome 9 (dark blue) and 1 chromosome 22 (red) in an interphase cell nucleus. Locations of the ABL and BCR genes are indicated by arrows; centromeres by contractions. If 2 DNA double-strand breaks occur, one in each of the 2 genes, and the breaks come close together spatially, misrepair can induce the translocation shown schematically in panel B. The translocation makes a BCR-ABL oncogene (too small to be visible at this scale) located at the color junction shown, that is, can turn a LSC-predecessor cell into a CML LSC. By modeling the BCR-ABL translocation frequency based on the sizes of the relevant introns of BCR and ABL, and comparing the resulting probability with CML incidence, it is possible to estimate the number of LSC-predecessor cells at risk, independently for radiation-induced and age-induced translocations. Experimental data show that in normal individuals, the 2 loci are on average closer together than randomness would indicate, implying a higher BCR-ABL translocation probability and thus a lower estimate of the number of LSC-predecessor cells needed to explain observed CML incidence.
Proximity effects. (A) Schematically shows 1 chromosome 9 (dark blue) and 1 chromosome 22 (red) in an interphase cell nucleus. Locations of the ABL and BCR genes are indicated by arrows; centromeres by contractions. If 2 DNA double-strand breaks occur, one in each of the 2 genes, and the breaks come close together spatially, misrepair can induce the translocation shown schematically in panel B. The translocation makes a BCR-ABL oncogene (too small to be visible at this scale) located at the color junction shown, that is, can turn a LSC-predecessor cell into a CML LSC. By modeling the BCR-ABL translocation frequency based on the sizes of the relevant introns of BCR and ABL, and comparing the resulting probability with CML incidence, it is possible to estimate the number of LSC-predecessor cells at risk, independently for radiation-induced and age-induced translocations. Experimental data show that in normal individuals, the 2 loci are on average closer together than randomness would indicate, implying a higher BCR-ABL translocation probability and thus a lower estimate of the number of LSC-predecessor cells needed to explain observed CML incidence.
Dose-response data support the proximity scenario of Figure 7, based on standard radiobiologic results for the relevant doses,31,78 as follows: (1) usually, 1-track radiation action produces a linear dose response and 2-track radiation action produces a quadratic dose-response; (2) translocations are produced by both 1-track and 2-track action, with the former dominating at lower and the latter dominating at higher doses50 ; and (3) if 2 loci are on average abnormally close together, then 1-track radiation action is enhanced, because of an increased probability that a single radiation track could cause both breaks with the resulting broken loci then so close in space and time that they can readily misrejoin with each other.81 Combining (1) through (3) shows that the proximity scenario leads to an enhancement of 1-track, dose-linear action. This result provides a reason why epidemiologic estimates of radiation-induced CML risks are approximately linear for doses < ∼ 4 Gy,48 even though a linear-quadratic dose response is predicted and observed for total chromosome translocations (reviewed in Edwards50 ), and a nearly quadratic dose response is observed for other radiation-induced leukemias.61 It is in such arguments that detailed biophysical knowledge about ionizing radiation assists mechanistic insights.
Importance
Whether the LSC-predecessor cells are HSCs is a key question, important to CML modeling, CML treatment planning, estimating risks of radiation-induced CML,87,88 and even stem cell research. The CML models17,39 differ widely in their estimates of HSC numbers, from ∼ 400 up to much higher values (> 106) for total (cycling plus resting) HSCs. The radiobiologic LSC-predecessor number estimates, even after corrections for proximity effects, are somewhat larger still.
If LSC-predecessor cells are HSCs, the results in “Radiobiologic and background data” are important for stochastic CML models' conceptually and clinically relevant predictions. In particular, predictions of what happens if TKI treatment is discontinued,7 interrupted, or poorly adhered to89 depend on the HSC number.
There is some evidence for a “dead-band” in the control of HSC numbers,90-93 as described in Radivoyevitch et al87 ; here “dead” refers to the putative nonresponsive character of the HSC feedback control system when HSC numbers are more than sufficient to maintain a hematopoietic system. This postulate is an example of the systems biology concept of bounded autonomy.94 If correct, the postulate would imply that there could be “CML prophylaxis by hematectomy” (ie, by reduction of HSC number) whereby losses of HSCs in individuals who have more than enough HSCs neither trigger repopulation nor compromise the function of the individual's hematopoietic system, and serendipitously are beneficial to the extent that LSC predecessors have decreased in number, thus lowering the individual's risk of CML. We suggest that enigmatic low background and radiation-induced CML incidence among Nagasaki atomic bomb survivors48 may be an example of such prophylaxis, that is, a highly prevalent pre-atomic bomb population stress may have substantially and systematically depleted this particular population of its HSC reserve.88
The dead-band hypothesis could help explain some details shown in Figure 6. If there is a dead-band, HSC losses with aging (because of random cell deaths) are more likely than gains, so that ∼ 2-fold HSCs decrease from 29 years and 48 years, corresponding to the CML incidence decrease shown in the high-dose curve, are not unreasonable. In any case, ∼ 2-fold increases in body mass between 11 and 29 year olds (and thus LSC-predecessor cell numbers), corresponding to the CML incidence increase shown in the curve, are also not unreasonable.
Conclusions
Updated radiobiologic data and modeling strongly indicate that the CML latency time distribution has an extended large-time tail. All current cell population dynamics CML models that deal with clinical (rather than epidemiologic) issues substantially underestimate the length of the tail. For CML treatment, one conclusion is that voluntary discontinuance, voluntary interruption, or poor patient adherence to protocol of TKI treatments should be regarded with caution, especially for females, at least pending the final outcome of the STIM7 and similar trials. Long and highly variable latency times suggest that unpredictable relapses could occur, even after decades. Moreover, for birth-death models of LSC proliferation, higher variance in latency time distribution for a given mean is correlated with a higher per cell death rate for a given net (birth minus death) per cell growth rate, so that on average more cell divisions are required to achieve a given total growth,41 which in turn means, according to the CML models,15,95 higher chances for TKI mutations before diagnosis. As regards risk estimation for radiogenic CML, the main implication is that more attention should be paid to the cryptic latency period between 2 comparatively well understood phases: radiation initiation of CML and postdiagnosis CML dynamics. CML latency time probability distributions are also important for the insights CML studies can give on fundamental cancer biology in general (reviewed in Dingli et al96 ).
For many years, quantitative explanations of age-driven increases in background tumor incidence during adulthood have been based on multistage models.66,67,77,97-99 Such models assume a constant incidence rate for oncogenic mutations (and for other alterations such as chromosome translocations) during adulthood. They attribute the increases in tumor incidence to clonal expansion and/or the need, at most tumor sites though not for CML, to accumulate more than one alteration (eg, 2 mutations and one translocation) in a single clone before malignancy occurs. Figure 5 and its analysis give evidence that assuming constancy of alteration incidence as age increases may be a poor approximation in some cases. We do not know how BCR-ABL data would compare with the data shown in Figure 5 for all translocations lumped together, nor do we know how relevant CML initiation data are to other cancers (1 other leukemia site is analyzed in supplemental Figure 4). But if increasing incidence of alterations during adulthood, synergistic with the mechanisms assumed in multistage models, is in fact significant for many tumor sites, a major change in carcinogenesis paradigms is called for. The implication for risk models for such sites would be a shift to alteration rate parameters that increase with age, and also to models with fewer stages. In any case, investigations of BCR-ABL incidence age dependence are indicated.
Chromosome translocations31 are important intermediates between DNA double-strand breaks and cancers, especially leukemias (reviewed in Mitelman et al100 ). Increasingly, clear understanding of age and dose responses of CML compared with BCR-ABL translocations should help clarify relationships between other cancers and their associated chromosomal translocations.
Estimates of the number of LSC predecessors vary by > 4 orders of magnitude (4 logs), with no clear explanation for such large discrepancies and with radiobiologic estimates favoring values so large as to cast some doubt on the assumption that LSC predecessors are HSC. Estimating the number of LSC predecessors is an area where increasingly accurate CML and radiobiologic models and data will inform and help correct each other.
Connections between disparate datasets can be made through mathematical models, and the synthesis of data in this manner can lead to inferences beyond those that can be made by analyzing individual datasets in isolation. One example discussed here was comparing the age and dose responses of CML to the corresponding responses of chromosomal translocations to form new estimates of LSC-predecessor numbers. In particular, CML age response and radiation-dose response understanding is synergistic. For example, in equation 1, background CML incidence model accuracy impacts radiation-induced CML model accuracy, as they are coupled through fits to the data.
As regards future modeling developments, understanding CML LSCs better is critical for fundamental mechanistic understanding of CML. For clinical applications (supplemental Section 1), important additional projects include better models for resistance mutations, progression, and the relation of post–BM transplantation data to data on initiation, latency, and TKI treatments. In addition, since radiobiology analyses suggest possible sex differences for CML, emphasizing stratification by sex may be useful in future CML clinical trials.
Increased cooperation is needed among mathematical modelers, cancer biologists, and clinicians because the range of data types and perspectives needed to develop and calibrate predictive CML models is so wide.21
The online version of this article contains a data supplement.
Acknowledgments
The authors are grateful to Clare Lamont for helping prepare the cover illustration.
T.R. is grateful for support from National Institutes of Health R01 CA1388-03 and the Department of Epidemiology and Biostatistics at Case Western Reserve University. R.K.S. was supported by National Cancer Institute Integrative Cancer Biology Program (NCI ICBP) U54CA149233-029689 and DE-SC0001434 Office of Science (Office of Biological and Environmental Research [BER]) US Department of Energy. L.H. was supported by DE-SC0002606 Office of Science (BER) US Department of Energy and NCI U54CA149233. J.L. was supported by a summer stipend from the University of California UG Research Apprentice Program and by NCI ICBP 5U54CA149233-029689.
This report makes use of data obtained from the Radiation Effects Research Foundation (RERF), Hiroshima and Nagasaki, Japan. RERF is a private, nonprofit foundation funded by the Japanese Ministry of Health, Labor and Welfare and the US Department of Energy, the latter through the National Academy of Sciences. The conclusions in this report are those of the authors and do not necessarily reflect the scientific judgment of RERF or its funding agencies.
National Institutes of Health
Authorship
Contribution: T.R., L.H., and R.K.S. planned the review, researched the literature, and wrote the review; R.K.S., and especially T.R., carried out computer calculations relating CML modeling to radiobiology; and J.L. designed, wrote, tested, and used the R script needed to analyze one of the CML models (supplemental Table 1 top line), and also critiqued the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dr Rainer K. Sachs, Department of Mathematics, University of California, Berkeley, Evans Hall, MC3840, Berkeley, CA 94720; e-mail: sachs@math.berkeley.edu.
