

Key Points
PolyP, like heparin, is a physiologic cofactor for the C1-INH:C1s interaction, and thus a key negative regulator of complement.
Colocalization of polyP with C1-INH in activated platelets suggests that its cofactor function is physiologically relevant.
Abstract
The complement system plays a key role in innate immunity, inflammation, and coagulation. The system is delicately balanced by negative regulatory mechanisms that modulate the host response to pathogen invasion and injury. The serpin, C1-esterase inhibitor (C1-INH), is the only known plasma inhibitor of C1s, the initiating serine protease of the classical pathway of complement. Like other serpin-protease partners, C1-INH interaction with C1s is accelerated by polyanions such as heparin. Polyphosphate (polyP) is a naturally occurring polyanion with effects on coagulation and complement. We recently found that polyP binds to C1-INH, prompting us to consider whether polyP acts as a cofactor for C1-INH interactions with its target proteases. We show that polyP dampens C1s-mediated activation of the classical pathway in a polymer length- and concentration-dependent manner by accelerating C1-INH neutralization of C1s cleavage of C4 and C2. PolyP significantly increases the rate of interaction between C1s and C1-INH, to an extent comparable to heparin, with an exosite on the serine protease domain of the enzyme playing a major role in this interaction. In a serum-based cell culture system, polyP significantly suppressed C4d deposition on endothelial cells, generated via the classical and lectin pathways. Moreover, polyP and C1-INH colocalize in activated platelets, suggesting that their interactions are physiologically relevant. In summary, like heparin, polyP is a naturally occurring cofactor for the C1s:C1-INH interaction and thus an important regulator of complement activation. The findings may provide novel insights into mechanisms underlying inflammatory diseases and the development of new therapies.
Introduction
C1-esterase inhibitor (C1-INH) is a member of the superfamily of serine protease inhibitors (serpins) that regulate complement, coagulation, and inflammation. In the complement system, C1-INH is the major negative regulator of the classical and lectin pathways. It inhibits serine proteases C1s and C1r in the classical pathway,1 and mannose-binding lectin (MBL)-associated serine proteases (MASP)-1 and -2 in the lectin pathway,2 interfering with cleavage of complement factor C4 and C4b-bound C2, and formation of the C3 convertase, C4b2a. C1-INH also inhibits coagulation factors XIa (FXIa)3,4 and FXIIa,5 and plasma kallikrein.6 Kallikrein amplifies the contact pathway of coagulation through cleavage/activation of FXII,7 and exhibits pro-inflammatory properties by promoting the generation of bradykinin and βFXIIa, the latter which may activate C1r.8 Deficiencies of C1-INH are manifest by hereditary angioedema,9 and genetic variants of the gene encoding C1-INH are associated with a heightened risk of age-related macular degeneration.10 C1-INH also has biological properties that extend beyond its protease inhibitory function.11 Plasma-derived and recombinant forms of C1-INH are approved to treat hereditary angioedema,12,13 have efficacy in several preclinical models of sepsis, inflammation, ischemia-reperfusion injury, and transplant rejection, and some evidence of benefit in humans.14,15
C1-INH is a soluble glycoprotein, circulating at a concentration of ∼2 to 4 μM. It is mainly synthesized in the liver, but also found in and secreted by endothelial cells,16 monocytes,17 and platelets.18,19 C1-INH comprises a heavily glycosylated N-terminal domain and a C-terminal serpin domain. Similar to the way in which the glycosaminoglycans (GAGs), heparin and heparan sulfate, bind to antithrombin and FXa or thrombin to augment neutralization of these coagulation enzymes,20 these GAGs also potentiate the function of C1-INH to neutralize several proteases, particularly C1s and MASP-2.3,21-24 Heparin, stored primarily in mast cell granules, is moderately more potent than heparan sulfate, which resides on the cell surface and in the subendothelial matrix.25 Heparin/heparan sulfate is the only known physiologic polyanion that modulates complement via an association with C1-INH, accelerating the interaction of C1-INH and C1s five- to 11-fold.26 Heparin also inhibits C1-INH neutralization of FXIIa and βFXIIa.27
Polyphosphate (polyP) is a naturally occurring, highly anionic linear polymer of monophosphate (monoP) units, linked by phosphoanhydride bonds. Expressed in all cells, the polymers vary in length from 30 to 800 monoP units in mammalian cells,28 to up to thousands of units in some bacteria.29 PolyP with a length of 60 to 100 monoP units,30,31 is abundant in dense granules of platelets, released upon activation.31 Mast cells also contain high concentrations of polyP.32 Several lines of evidence indicate that polyP triggers the contact pathway of coagulation, and promotes coagulation at multiple steps in the cascade.33-37 We recently explored the role of polyP in the complement system and showed that it has profound suppressive effects on the terminal pathway.38 We also found that polyP binds to C1-INH, raising the possibility that polyP, like heparin/heparan sulfate, may act as a cofactor for C1-INH, augmenting its capacity to dampen complement activation.
In this report, we show that polyP is a physiologic polyanionic cofactor for C1-INH. PolyP potentiates the inhibitor function of C1-INH in a concentration- and size-dependent manner, dampening complement activation by augmenting C1-INH–mediated interference of C1s cleavage of C4 and C2. With similar kinetics, sites of interaction and specificity as heparin, polyP interacts with C1s and C1-INH, promoting rapid formation of a serpin-protease complex. The potential relevance of these findings is underscored by the finding that polyP can reduce C4d deposition on cultured endothelial cells via interference of the classical and lectin pathways, and that polyP and C1-INH colocalize in activated human platelets. These findings provide new and potentially clinically important insights into the mechanisms by which complement is regulated.
Materials and methods
Effect of polyP on C1-INH inhibition of C1s-mediated cleavage of C2 and C4
PolyP or monoP was pre-incubated with C1-INH for 60 minutes at 37°C in a buffer containing 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.5, 150 mM NaCl, and 0.01% Tween 20. C1s was added for 30 minutes, followed by C4 or C2. The reaction was allowed to proceed for 5 minutes and stopped with Laemmli buffer. Samples were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and protein bands were visualized with Coomassie blue R-250.
Enzyme kinetics
The C1s–C1-INH interaction was measured by an absorbance-based assay employing Z-Lys-SBzl as the substrate and the chromogenic thiol reagent 4,4′-dithiodipyridine (DTDP).39,40 Residual C1s activity was monitored at 324 nm or 344 nm. Assays were conducted in 20 mM Tris, 100 mM NaCl, 0.005% Triton X-100, 5% dimethyl sulfoxide, pH 7.4 at 25°C in a Cary 4000 UV-Visible spectrophotometer (Varian) or a FLUOstar Omega plate reader (BMG Labtech). Progress curves were fitted by nonlinear regression using OriginPro 9 (OriginLab Corporation) to the integrated rate equation for slow binding inhibition.41,42
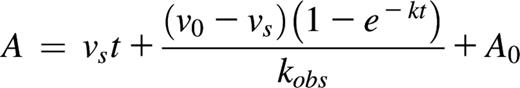
C4d deposition on endothelial cells
Human dermal microvascular endothelial cells (HMEC-1)43 were washed with phosphate-buffered saline (PBS), dissociated with accutase, washed and suspended in 100 μL PBS at a concentration of ∼8 × 106 cells/mL. Cells were incubated at 37°C for 1 hour with 10% normal human serum (NHS), C1q-depleted serum, MBL-deficient serum, or corresponding serum that was complement-inactivated using heat (1 hour at 57°C). Cells were washed and resuspended in 75 μL of fluorescence-activated cell sorter (FACS) buffer (PBS, containing 1% [weight-to-volume ratio] bovine serum albumin) with murine monoclonal anti-C4d antibody 4 μg/mL for 1 hour at 4°C, pelleted and resuspended in 100 μL FACS buffer with polyclonal fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin G (1:400; ∼5 μg/mL) for 45 minutes at 4°C. Cells were finally suspended in 500 μL FACS buffer with 0.1 mg/mL propidium iodide and analyzed by flow cytometry using a FACS LSR system (BD Biosciences, Mountain View, CA).
Immunolocalization of C1-INH and polyP in platelets
Research involving human subjects was approved by the University of British Columbia Clinical Research Ethics Board. Human platelets from platelet-rich plasma were prepared as reported.44 Resting platelets were activated with the phorbol 12-myristate 13-acetate (100 nM) and A23187 (1 μM), plated onto glass coverslips, fixed with 2% paraformaldehyde, permeabilized with 0.1% Triton-X-100, and blocked with 1% bovine serum albumin. Platelets were then incubated with anti–C1-INH primary antibody (LSBio, Seattle, WA) for 1 hour at room temperature, followed by the fluorescein isothiocyanate-conjugated secondary antibody for 1 hour. PolyP was labeled with a mixture of biotinylated polyP binding domain (PPXbd)45 and tetramethylrhodamine-conjugated streptavidin (DyLight; Life Technologies, Grand Island, NY). Confocal images were processed using a Zeiss spinning disk confocal microscope and SlideBook software (Intelligent Imaging Innovations, Denver, CO). Images supplied as grayscale were colorized in green or red using Adobe Photoshop.
Statistics
Unpaired Student t tests were performed with GraphPad Prism version 5 software (San Diego, CA). Studies were performed in triplicate unless otherwise noted. Significance: P < .05.
Reagents, analytical chromatography, and surface plasmon resonance (SPR)
See supplemental Data in corresponding sections, available on the Blood Web site.
Results
PolyP potentiates C1-INH inhibition of C1s cleavage of C4 and C2 in a concentration-dependent manner
The effect of polyP on the capacity of C1-INH to neutralize C1s was first evaluated by monitoring C1s-mediated cleavage of C4 in gel-based assays in the presence of varying concentrations of polyP130. This size was considered reasonable to study, as polyP12531 and polyP13038 exhibited similar effects as the slightly shorter platelet-size polyP.
At concentrations above 50 to 100 μM (concentrations of polyP reported herein are based on the monoP [NaPO3] units; thus true concentrations of the polymer can be derived by dividing reported concentrations by the number of monoP units), the addition of polyP130 to fixed concentrations of C1-INH, C1s, and C4, dampened C1s-mediated C4 cleavage to C4a and C4b, monitored by proteolysis of the C4 α-chain to the C4 α′-chain (Figure 1A). Cleavage was progressively dampened as the polyP130 concentration was increased to 500 μM. At 500 μM, monoP had no effect on C1s-mediated cleavage of C4 with the same concentration of C1-INH (5th lane). Thus, the polymer form of phosphate is required to potentiate the function of C1-INH.

PolyP enhances the capacity of C1-INH to dampen C1s-mediated cleavage of C4 and C2 in a concentration- and size-dependent manner. SDS-PAGE analysis under reducing conditions was used to evaluate the effect of polyP concentration and size, on C4 and C2 cleavage. Concentrations of polyP reflect the concentration of the monomer. (A) C4 alone is shown in lane 1. C1s alone is not detectable at the concentration used. C1-INH and C1s were reacted with C4 in the presence of varying concentrations of polyP130 or 500 μM of monoP (lane 5) as shown and described in supplemental Materials and methods. C4 cleavage products were detected by Coomassie staining after SDS-PAGE. (B) C1-INH and C1s were reacted with C2 in the presence of varying concentrations of polyP130 or 500 μM of monoP (lane 5). (C) C1-INH and C1s were reacted with C4 in the presence of 500 μM monoP (P1), P3, polyP14 (P14), polyP60 (P60), or polyP130 (P130) for 30 minutes, after which C4 cleavage products were visualized by SDS-PAGE. Gels are representative of experiments performed a minimum of 5 times.
PolyP enhances the capacity of C1-INH to dampen C1s-mediated cleavage of C4 and C2 in a concentration- and size-dependent manner. SDS-PAGE analysis under reducing conditions was used to evaluate the effect of polyP concentration and size, on C4 and C2 cleavage. Concentrations of polyP reflect the concentration of the monomer. (A) C4 alone is shown in lane 1. C1s alone is not detectable at the concentration used. C1-INH and C1s were reacted with C4 in the presence of varying concentrations of polyP130 or 500 μM of monoP (lane 5) as shown and described in supplemental Materials and methods. C4 cleavage products were detected by Coomassie staining after SDS-PAGE. (B) C1-INH and C1s were reacted with C2 in the presence of varying concentrations of polyP130 or 500 μM of monoP (lane 5). (C) C1-INH and C1s were reacted with C4 in the presence of 500 μM monoP (P1), P3, polyP14 (P14), polyP60 (P60), or polyP130 (P130) for 30 minutes, after which C4 cleavage products were visualized by SDS-PAGE. Gels are representative of experiments performed a minimum of 5 times.
Similar to C1s cleavage of C4, polyP130 in excess of 25 to 50 μM enhanced the inhibitory properties of C1-INH on C1s-mediated cleavage of C2 (Figure 1B). Again, monoP did not alter C1s-mediated cleavage of C2 in the presence of C1-INH (5th lane).
Enhancement of the inhibitory properties of C1-INH by polyP is dependent on its size
The effects of polyP on coagulation and the terminal pathway of complement are size dependent, ie, longer polymers are more potent.35,38 We assessed the size-dependence of polyP on the capacity of C1-INH to neutralize C1s-mediated cleavage of C4 (Figure 1C). At an equivalent molar concentration of polyP based on the monomeric form, P1 and P3 had no effect on C1s cleavage of C4 in the presence of C1-INH. However, polyP, equal to or greater in length than P14, was increasingly effective in potentiating C1-INH neutralization of C1s-mediated cleavage of C4. This was not due to chelation of cations that may have been present, because the addition of 5 mM EDTA to the reaction did not alter the results (not shown).
Heparin and polyP increase the rate of complex formation between C1s and C1-INH
To demonstrate that polyP was mediating its effects on the reaction between C1s and C1-INH by increasing the rate of the canonical complex formation between the two molecules, the rate of complex formation was measured using SDS-PAGE analysis (Figure 2A). This takes advantage of the formation of a covalent complex between the serpin and C1s. In the presence of polyP130 at 16 and 166 μM, the formation of the C1s:C1-INH complex was visualized essentially instantaneously (0 minutes), despite the reaction being conducted on ice. Formation of the complex was concomitant with a reduction in the intensity of the serine protease (SP) domain of C1s (C1s SP domain). A lower concentration of polyP130 (3 μM) did not have as great an effect, ie, it was similar to the reaction in the absence of polyP130 (Figure 2A, lanes 1-2 of left panel). After 1 minute, the difference between the reactions in the presence of higher concentrations of polyP130 was still notable, with the highest concentration having the greatest effect. After 5 minutes, all reactions were essentially complete.

Heparin and polyP increase the rate of complex formation between C1-INH and C1s. For panels A-D, proteins were separated by 10% SDS-PAGE and gels were stained with Coomassie blue R-250. Concentrations of polyP reflect the concentration of the monomer. (A) 1 μM C1-INH was reacted at 4°C with 1 μM recombinant C1s (C1s SP domain), in the presence of increasing concentrations of polyP130 (P130) for the indicated times. Purified C1-INH is shown in the lane between the 1 and 5 minute panels. (B) 1 μM C1-INH was reacted at 4°C for 0 to 30 minutes with 1 μM recombinant C1s (C1s SP domain) in the absence (C, control) or presence of either 50 μg/mL heparin or 160 μM polyP130 (left). Densitometry performed on 3 gels (right). The percent C1-INH complex is based on C1-INH alone being assigned as 100%. (C) 1 μM C1-INH was reacted at 4°C for 0 to 120 minutes with 1 μM plasma C1r in the absence (C) or presence of either 50 μg/mL heparin or 166 μM polyP130 (left). Densitometry performed on 3 gels (right). The percent C1-INH complex is based on C1-INH at t = 0 being assigned as 100%. (D) 10 nM C1-INH was reacted at 4°C for varying periods of time as shown, with 10 nM C1s (CCP12SP), 1 μM C4, and either buffer alone (C), 50 μg/mL heparin, or 166 μM polyP130 (left). Densitometry performed on 3 gels (right). The percent C4 α chain is based on C4α at 5 hours being assigned as 100%. CCP, complement control protein-like domain; H, heparin; P, polyP130.
Heparin and polyP increase the rate of complex formation between C1-INH and C1s. For panels A-D, proteins were separated by 10% SDS-PAGE and gels were stained with Coomassie blue R-250. Concentrations of polyP reflect the concentration of the monomer. (A) 1 μM C1-INH was reacted at 4°C with 1 μM recombinant C1s (C1s SP domain), in the presence of increasing concentrations of polyP130 (P130) for the indicated times. Purified C1-INH is shown in the lane between the 1 and 5 minute panels. (B) 1 μM C1-INH was reacted at 4°C for 0 to 30 minutes with 1 μM recombinant C1s (C1s SP domain) in the absence (C, control) or presence of either 50 μg/mL heparin or 160 μM polyP130 (left). Densitometry performed on 3 gels (right). The percent C1-INH complex is based on C1-INH alone being assigned as 100%. (C) 1 μM C1-INH was reacted at 4°C for 0 to 120 minutes with 1 μM plasma C1r in the absence (C) or presence of either 50 μg/mL heparin or 166 μM polyP130 (left). Densitometry performed on 3 gels (right). The percent C1-INH complex is based on C1-INH at t = 0 being assigned as 100%. (D) 10 nM C1-INH was reacted at 4°C for varying periods of time as shown, with 10 nM C1s (CCP12SP), 1 μM C4, and either buffer alone (C), 50 μg/mL heparin, or 166 μM polyP130 (left). Densitometry performed on 3 gels (right). The percent C4 α chain is based on C4α at 5 hours being assigned as 100%. CCP, complement control protein-like domain; H, heparin; P, polyP130.
The effects of polyP130 and heparin on C1s:C1-INH complex formation were compared (Figure 2B). Complex formation, concomitant with reduced intensity of the C1s SP band, was essentially instantaneous (0 minutes) with either heparin or polyP130, indicating that the cofactors had similar effects. The difference made by the cofactors could also be visualized after 0.5 and 1 minute, but at 3 minutes and thereafter, all reactions were essentially complete. Neither polyP130 nor heparin exhibited differential effects on complex formation between C1-INH and C1r, indicating that C1s is the primary target of these polyanions (Figure 2C).
Importantly, no free cleaved C1-INH could be detected using this analysis, indicating polyP130 and heparin did not affect the stoichiometry of the serpin-enzyme reaction.
Based on the similar kinetics of C1s:C1-INH complex formation potentiated by polyP130 or heparin, we predicted that these anions would similarly augment the capacity of C1-INH to interfere with C1s cleavage of its substrates. This was validated by gel-based assays and confirmed by densitometry, in which C1s was incubated with C4 in the presence of C1-INH (Figure 2D). The appearance of the C4 α′-chain fragment was evidence of C4 cleavage by C1s. Without heparin or polyP130 (first lane of each panel noted with C [control]), it took over 90 minutes for C1-INH to suppress C1s-mediated cleavage of C4. In contrast, heparin and polyP130 enhanced the rate of inhibition of C1s cleavage of C4, such that proteolytic products disappeared within 5 to 15 minutes of incubation. Notably, heparin and polyP130 exhibited similar kinetic profiles.
Heparin and polyP130 increase the rate of interaction between C1-INH and C1s
The effect of polyP130 on the observed rate of interaction (kobs) between C1-INH and C1s, in comparison with heparin, was measured using the enzyme activity against a peptidyl substrate as readout of the progress of the inhibition (Figure 3A). PolyP130 increased the kobs for association of C1-INH and wild-type (WT) recombinant C1s by ∼90-fold at concentrations above 1000 μM. This enhanced rate was not seen for a mutant of C1s (A1) in which the 4 positively charged residues constituting an exosite on the surface of the enzyme46,47 were mutated to neutrally charged alanine residues (Figure 3B). Thus, the exosite of C1s, an accessory-binding site for the C4 substrate of the enzyme,46 also constitutes an important site for interactions with polyP130. Similar results were found for heparin, with concentrations above 20 μg/mL, increasing the rate of association between WT C1s and C1-INH ∼90-fold (Figure 3C), an effect not seen for the C1s A1 mutant (Figure 3D). Therefore, heparin and polyP have similar modes of action.

Effect of polyP130 and heparin on the observed rates of association between C1s and C1-INH. The effect of polyP130 (A) or heparin (C) on the observed rate of association between recombinant WT CCP12SP C1s and C1-INH was determined by adding C1s (2 nM) to C1-INH (20 nM), Z-Lys-SBzl (0.2 mM), and DTDP (0.6 mM) in the presence of increasing concentrations of polyP130 (0-2000 μM) (A) or heparin (0-100 μg mL−1) (C). The pseudo-first-order rate constant (kobs) is plotted as a function of polyP130 or heparin concentration. The right-hand axis shows the ratio of the kobs at a particular polyP130 or heparin concentration to that in the absence of a cofactor. The effect of polyP130 (B) or heparin (D) on the observed rate of association between recombinant exosite (A1) mutant CCP12SP C1s and C1-INH was determined by adding 1.5 nM C1s A1 mutant to 30 nM C1-INH, 0.2 nM Z-Lys-SBzl, and 0.9 mM DTDP in the presence of increasing concentrations of polyP130 (0-2000 μM) (B) or heparin (0-500 μg mL−1) (D). The pseudo-first-order rate constant (kobs) is plotted as a function of polyP130 or heparin concentration. The right-hand axis shows the ratio of the kobs at a particular polyP130 or heparin concentration to that in the absence of a cofactor. Results are representative of 3 independent experiments.
Effect of polyP130 and heparin on the observed rates of association between C1s and C1-INH. The effect of polyP130 (A) or heparin (C) on the observed rate of association between recombinant WT CCP12SP C1s and C1-INH was determined by adding C1s (2 nM) to C1-INH (20 nM), Z-Lys-SBzl (0.2 mM), and DTDP (0.6 mM) in the presence of increasing concentrations of polyP130 (0-2000 μM) (A) or heparin (0-100 μg mL−1) (C). The pseudo-first-order rate constant (kobs) is plotted as a function of polyP130 or heparin concentration. The right-hand axis shows the ratio of the kobs at a particular polyP130 or heparin concentration to that in the absence of a cofactor. The effect of polyP130 (B) or heparin (D) on the observed rate of association between recombinant exosite (A1) mutant CCP12SP C1s and C1-INH was determined by adding 1.5 nM C1s A1 mutant to 30 nM C1-INH, 0.2 nM Z-Lys-SBzl, and 0.9 mM DTDP in the presence of increasing concentrations of polyP130 (0-2000 μM) (B) or heparin (0-500 μg mL−1) (D). The pseudo-first-order rate constant (kobs) is plotted as a function of polyP130 or heparin concentration. The right-hand axis shows the ratio of the kobs at a particular polyP130 or heparin concentration to that in the absence of a cofactor. Results are representative of 3 independent experiments.
The positively charged exosite on the C1s SP domain contributes to binding of polyP and heparin
Using analytical heparin-affinity chromatography (supplemental Data, see “Analytical chromatography”), WT C1s (CCP12SP) (Figure 4A, solid black line) eluted off the column the latest, and thus binds more strongly to heparin than the C1s A1 mutant (orange line) or C1-INH (gray line). We speculate that the C1s A1 mutant eluted in multiple peaks due to the protein assuming multiple conformations with differing heparin affinity in solution.47 As expected, the C1s:C1-INH complex bound to the matrix with an affinity, intermediate between the inhibitor and the enzyme. This indicates that the enzyme’s SP exosite is important for interactions with heparin, although other residues of the enzyme also likely participate in binding to heparin, as binding was not ablated for the C1s A1 mutant.

Interactions of C1-INH and C1s with heparin and polyP. (A) Analytical affinity chromatography indicates binding of heparin to C1s and C1-INH. C1-INH (gray line), plasma-derived C1s (black line), a 1:1 mixture of both (pre-incubated for 60 minutes at 37°C) (dotted black line), or the C1s A1 mutant (orange solid line), were separately applied to a HiTrap-Heparin column and eluted with a NaCl gradient as described in “Materials and methods.” Conductivity measurements are shown on the right-hand axis. Eluted fractions were analyzed by SDS-PAGE to confirm the identity of the proteins. (B) SPR was used to quantify binding of C1s (CCP12SP) to biotinylated polyP250 attached to streptavidin immobilized to the chip. A representative experiment (n = 3) shows curves for 0 to 625 nM C1s flowed over the immobilized polyP250. Inset: The response units obtained at equilibrium for each concentration of C1s were plotted and fitted using a one-site binding model on GraphPad Prism (regression coefficient = 0.99). Similar results were obtained in 2 additional independent experiments.
Interactions of C1-INH and C1s with heparin and polyP. (A) Analytical affinity chromatography indicates binding of heparin to C1s and C1-INH. C1-INH (gray line), plasma-derived C1s (black line), a 1:1 mixture of both (pre-incubated for 60 minutes at 37°C) (dotted black line), or the C1s A1 mutant (orange solid line), were separately applied to a HiTrap-Heparin column and eluted with a NaCl gradient as described in “Materials and methods.” Conductivity measurements are shown on the right-hand axis. Eluted fractions were analyzed by SDS-PAGE to confirm the identity of the proteins. (B) SPR was used to quantify binding of C1s (CCP12SP) to biotinylated polyP250 attached to streptavidin immobilized to the chip. A representative experiment (n = 3) shows curves for 0 to 625 nM C1s flowed over the immobilized polyP250. Inset: The response units obtained at equilibrium for each concentration of C1s were plotted and fitted using a one-site binding model on GraphPad Prism (regression coefficient = 0.99). Similar results were obtained in 2 additional independent experiments.
We measured the binding properties of C1s and C1s A1 mutant to polyP in 2 ways. First, we used SPR to characterize the binding of C1s to immobilized biotinylated polyP25048 (supplemental Data; Figure 4B). Fitting of the data to a steady state analysis yielded a KD of 198 nM. Second, we used biolayer interferometry with an Octet Red instrument and determined that the KD for C1s binding to biotinylated polyP130 was 286 nM (R2 = 0.986) (data not shown). Thus, the interaction of WT C1s with polyP250 and polyP130 is similar, and most notably, these are similar to what was reported for C1s with heparin (KD = 488 nM).26 Binding of the C1s A1 mutant to polyP250 was evaluated using SPR as for the WT enzyme, yielding a KD of 778 nM, indicating that the SP domain exosite of the enzyme is important for the binding of polyP by C1s in a similar manner to what was found for heparin. Using SPR, we also characterized C1-INH binding to immobilized biotinylated polyP250, and calculated a KD of 450 nM (supplemental Figure 1). The KD for C1-INH binding to heparin is 167 nM,49 again showing that polyP and heparin bind C1-INH similarly.
Inhibition of complement activation on endothelial cells exposed to polyP130
To evaluate the potential physiologic relevance of the interaction of polyP with C1-INH, we first examined the effect of polyP on complement activation via the classical and lectin pathways in a serum system using HMEC-143 as a target. Flow cytometry was used to measure deposition of C4d that is generated by FI-mediated cleavage of C4b, subsequent to formation of the classical/lectin pathway C3 convertase. With NHS (Figure 5A), C4d deposition was readily detected, whereas the addition of heat-inactivated serum resulted in minimal C4d deposition (Figure 5A, right panel). When cells were exposed to NHS in the presence of the highest concentration of monoP (1 mM), C4d deposition did not appreciably change. However, polyP130 caused a concentration-dependent reduction (P < .05 for ≥125 μM) in C4d deposition (Figure 5A). Under the same conditions, and as expected,33 polyP130 activated FXII (data not shown), which may result in C1r activation via βFXIIa8 and thereby, in contrast, activate complement via the classical pathway. However, the over-riding effect of polyP130 in this serum-based cell culture system was to dampen C4 cleavage, likely by potentiating the protease inhibitory properties of C1-INH.

PolyP inhibits C4d deposition on endothelial cells in a concentration-dependent manner. HMEC-1 were exposed to (A) NHS, (B) MBL-deficient serum, or (C) C1q-deficient serum in the presence of varying concentrations of polyP130 (P130) or monoP (P1). For each serum, a corresponding HIS control was tested (flow cytometry graphics on right panels). After 1 hour, the reactions were stopped, and C4d deposition was quantified by flow cytometry. Data shown are representative of 3 independent experiments for each serum condition. Histograms on the left show normalized MFI levels of C4d deposited on endothelial cells. Percent MFI values are mean ± SEM of duplicates. On the right, flow cytometry graphic reveals inhibition of C4d deposition, reflected by reduced fluorescence. HIS, heat-inactivated serum; MFI, mean fluorescence intensity; SEM, standard error of the mean.
PolyP inhibits C4d deposition on endothelial cells in a concentration-dependent manner. HMEC-1 were exposed to (A) NHS, (B) MBL-deficient serum, or (C) C1q-deficient serum in the presence of varying concentrations of polyP130 (P130) or monoP (P1). For each serum, a corresponding HIS control was tested (flow cytometry graphics on right panels). After 1 hour, the reactions were stopped, and C4d deposition was quantified by flow cytometry. Data shown are representative of 3 independent experiments for each serum condition. Histograms on the left show normalized MFI levels of C4d deposited on endothelial cells. Percent MFI values are mean ± SEM of duplicates. On the right, flow cytometry graphic reveals inhibition of C4d deposition, reflected by reduced fluorescence. HIS, heat-inactivated serum; MFI, mean fluorescence intensity; SEM, standard error of the mean.
Generation of C4d does not distinguish the classical from the lectin pathway. Participation of the classical pathway was confirmed using MBL-deficient serum (Figure 5B). Although monoP had a minimal effect, polyP130 markedly suppressed C4d deposition in a dose-dependent manner, with ∼50% suppression (P < .05) observed at a concentration of ∼100 μM polyP130. Interestingly, we observed similar results when we used C1q-deficient serum (Figure 5C) (P < .05 for concentrations ≥125 μM), suggesting that polyP130 also interferes with the lectin pathway.
PolyP and C1-INH colocalize in activated platelets
We also assessed the potential relevance of the interaction between C1-INH and polyP by examining their spatial distributions in platelets, where both are highly expressed, but housed in different organelles. A biotinylated recombinant yeast PPXbd45 was used to detect polyP. The specificity of PPXbd-polyP interaction was confirmed by the loss of a signal when platelets were incubated with a yeast exopolyphosphatase known to degrade polyP50 (not shown). C1-INH and polyP appeared to be separately distributed in resting platelets (Figure 6, top panels). In contrast, polyP and C1-INH appeared to coalesce and colocalize toward the centers of the activated platelets (Figure 6, bottom panels).

PolyP and C1-INH colocalize in activated human platelets. Human platelets were plated onto glass coverslips in the resting state (top panels) or after 2 minutes of activation with 100 nM of the phorbol ester, phorbol 12-myristate 13-acetate, and 1 μM of calcium ionophore A23187. After fixation and permeabilization, the platelets were stained as described in “Materials and methods” to detect C1-INH (left panels, green) and polyP (middle panels, red) by confocal microscopy. The right panels reveal the merged images. In resting platelets, C1-INH is detected in α granules and on the cell surface, and polyP is found in dense granules. After activation, C1-INH and polyP coalesce in the center of the platelets where they colocalize (yellow, right bottom panel). Size bars, 10 μm. Results are representative of 3 independent experiments.
PolyP and C1-INH colocalize in activated human platelets. Human platelets were plated onto glass coverslips in the resting state (top panels) or after 2 minutes of activation with 100 nM of the phorbol ester, phorbol 12-myristate 13-acetate, and 1 μM of calcium ionophore A23187. After fixation and permeabilization, the platelets were stained as described in “Materials and methods” to detect C1-INH (left panels, green) and polyP (middle panels, red) by confocal microscopy. The right panels reveal the merged images. In resting platelets, C1-INH is detected in α granules and on the cell surface, and polyP is found in dense granules. After activation, C1-INH and polyP coalesce in the center of the platelets where they colocalize (yellow, right bottom panel). Size bars, 10 μm. Results are representative of 3 independent experiments.
Discussion
We previously reported that polyP dampens complement activation via the terminal pathway by destabilizing C5b,6, and that this occurs in a size- and concentration-dependent manner.38 In this report, we show that polyP also suppresses complement activation via the classical pathway, potentiating the inhibitory function of the serpin, C1-INH, thereby interfering with C1s-mediated cleavage/activation of C4 and C2 (Figure 7). This effect of polyP on the function of C1s as regulated by C1-INH, also depends on the size and concentration of the polyanion, with polymers comprising more than 14 monoP units being increasingly more potent. Analogous to the effect of heparin/heparan sulfate on the interaction of antithrombin with thrombin or FXa, we found that polyP binds to both C1s and C1-INH, rapidly inducing the formation of a serpin-protease complex. The kinetics of complex formation and of inhibition of C1s cleavage of C4, were remarkably similar to that induced by heparin. We also demonstrated the potential physiologic relevance of our findings by 2 approaches: (1) the addition of polyP130 to serum dampens complement activation via the classical and lectin pathways with reduced generation and deposition of fragment C4d on endothelial cells, over-riding any potential complement-activating effects that polyP might mediate via activation of FXII, and (2) the colocalization of polyP and C1-INH in activated human platelets.

Mechanisms by which polyP regulates complement activation. Upon activation, polyP and C1-INH can colocalize, after which they are secreted. PolyP triggers a conformational change in FXII, resulting in generation of FXIIa, which can activate prekallikrein and/or FXI to FXIa. Kallikrein or plasmin (not shown) can further cleave FXIIa to generate βFXIIa, which may activate C1r and thus promote complement activation. C1-INH dampens that pathway by inhibiting FXIIa, βFXIIa, and kallikrein. C1s cleaves C4 and C2 to generate the C4b2a C3 convertase, which ultimately leads to formation of the C5b,6 complex, and assembly of the C5b-9 MAC. PolyP or heparin potentiate the inhibitory function of C1-INH via direct interactions with C1-INH and the target protease, C1s. PolyP also destabilizes C5b,6, thereby dampening the formation of the MAC. The over-riding effect of polyP in a serum-based endothelial cell culture system is to suppress complement activation. K, kallikrein; MAC, membrane attack complex; PK, prekallikrein.
Mechanisms by which polyP regulates complement activation. Upon activation, polyP and C1-INH can colocalize, after which they are secreted. PolyP triggers a conformational change in FXII, resulting in generation of FXIIa, which can activate prekallikrein and/or FXI to FXIa. Kallikrein or plasmin (not shown) can further cleave FXIIa to generate βFXIIa, which may activate C1r and thus promote complement activation. C1-INH dampens that pathway by inhibiting FXIIa, βFXIIa, and kallikrein. C1s cleaves C4 and C2 to generate the C4b2a C3 convertase, which ultimately leads to formation of the C5b,6 complex, and assembly of the C5b-9 MAC. PolyP or heparin potentiate the inhibitory function of C1-INH via direct interactions with C1-INH and the target protease, C1s. PolyP also destabilizes C5b,6, thereby dampening the formation of the MAC. The over-riding effect of polyP in a serum-based endothelial cell culture system is to suppress complement activation. K, kallikrein; MAC, membrane attack complex; PK, prekallikrein.
We therefore propose that polyP is a physiologic polyanion that, like heparin/heparan sulfate, accelerates C1-INH–mediated neutralization of the target protease, C1s. Indeed, our data reveal several similarities between heparin and polyP in terms of the functional relationship with C1-INH and C1s. C1-INH inhibits C1s by binding and dissociating the protease from the C1 complex, whereupon the complex is cleared from the circulation.51 Heparin is known to accelerate the serpin-protease interaction,52 probably by a so-called “charge sandwich mechanism,” whereby the polyanion heparin interposes as a negatively charged bridge between positively charged surfaces of C1-INH and C1s.24,53 Supportive of this concept, heparin oligomers containing 6 to 8 saccharide units were sufficient to achieve increases in the rate of inhibition.49 Our data support the notion that polyP and heparin bind, in part, to the positively charged exosite within the SP domain to enhance complex formation between C1-INH and C1s, and that the kinetics of the interactions are similar. PolyP lengths as short as 14 phosphate units were sufficient to achieve an effect, also consistent with a “charge sandwich mechanism,” rather than the template bridging mechanism by which heparin catalyzes antithrombin neutralization of thrombin or FXa.20 Notably, our studies provide the first confirmation that the positively charged exosite on the SP domain of C1s plays a direct role in interactions with heparin, a mechanism that applies also to the interaction of C1s with polyP. This interaction between the positively charged exosite on the C1s SP domain and heparin/polyP plays a pivotal role in enhancing the rate of interaction between the enzyme and C1-INH, because a mutant in which this exosite was mutated no longer displays an enhanced rate of interaction with C1-INH in the presence of the polyanions.
It is interesting that neither heparin nor polyP enhanced the rate of formation of C1-INH:C1r complexes in our gel-based assays. The findings, which are in line with the minimal effect that heparin has on the reaction between C1-INH and C1r,54 again underline the similarity between polyP and heparin, in terms of specificity. As mentioned, C1-INH also inhibits FXIa, FXIIa, βFXIIa,55 kallikrein, and MASP-1 and -2. Heparin, however, has little potentiating effect on C1-INH inhibition of kallikrein or MASP-1, and indeed dampens C1-INH neutralization of FXIIa.27 Heparin does however, augment C1-INH inhibition of FXIa and MASP-2.56 With respect to the latter, it is intriguing that polyP suppressed lectin pathway mediated C4 proteolysis and C4d deposition on endothelial cells, suggesting that polyP may also regulate MASP-2 by potentiating the inhibitory function of C1-INH. However, initial data (L.C.W., E.M.C., and R.N.P., unpublished data, January 2016) examining the effect of polyP on the interaction between MASP-2 and C1-INH does not support this being the mechanism whereby polyP exerts its effect on activation of the lectin pathway. Further studies to elucidate this effect are underway.
In spite of the above similarities, it is worth noting that there remain distinct differences in specificity between polyP and heparin. Indeed, heparin is an important cofactor for antithrombin-mediated neutralization of thrombin, accelerating the association >2000-fold.20 In contrast, polyP has no effect on the interaction between antithrombin and thrombin.33
What is the physiologic role of polyP, particularly in the context of C1-INH? We propose that under baseline conditions, the interaction between C1-INH and either heparin/heparan sulfate or polyP, provides continuous low-level surveillance in the blood, and protection against underlying tissue damage. C1-INH is synthesized by endothelial cells, where it is found on the cell surface, and likely helps to modulate the complement system and the contact system of plasma proteolysis on the vascular surface.16 Endothelial cells display abundant GAGs on their surface as heparan sulfate. PolyP derived from cellular sources, has been found at low concentrations in the blood of healthy individuals, circulating with a short half-life and lengths of 20 to 50 monoP units.57,58 The polyP would be available to bind to C1-INH on the endothelium, where it and/or heparan sulfate would enhance the function of C1-INH, allowing the C1-INH:polyanion complex to recruit and neutralize target proteases (eg, C1s). Interestingly, in our studies, pre-incubation of C1-INH with polyP was necessary to achieve maximal dampening of C1s cleavage of C4 and C2 (data not shown). This is similar to the requirement that heparin must first bind to C1-INH, for the serpin to accelerate association with C1s.26 Binding of the polyanion first to C1-INH would neutralize its surface charge, thereby allowing activated C1s to more rapidly and avidly bind to the serpin. This logical order of events would serve to limit the inflammatory response on the endothelial surface by continuously keeping complement activation in check. It is worth noting that in our studies, we compared polyP with heparin, showing essentially equal potency in accelerating the inhibitory function of C1-INH. However, it is heparan sulfate that is probably physiologically relevant in terms of vascular protection, and this GAG is less effective in enhancing C1-INH function than heparin.25 It therefore follows that polyP may actually be the prime accelerator of C1-INH function in vivo and be more important than heparan sulfate.
The protection of endothelial cells afforded by polyP interactions with C1-INH may extend to the surface of other cells. PolyP is prominently found in the dense granules of platelets and released upon activation.30,31 C1-INH is also found in platelets, but packaged in α-granules, from where it can be secreted and expressed on the activated platelet membrane.18 Although polyP and C1-INH are in discrete granules in the resting state, activation results in migration of granules toward the cell center, where they can fuse with each other and/or with the open canalicular system, resulting in secretion or delivery to the cell membrane.59,60 Colocalization of polyP and C1-INH in activated platelets suggests that these molecules can interact, and may thus collectively dampen complement activation on the host cell surface (Figure 7). It is reasonable to consider that disturbances in this process might result in disease. In that respect, patients with dense granule storage pool diseases, such as Hermansky-Pudlak syndrome,61 have low platelet levels of polyP,62 which may partly explain their tendency to bleed.31 However, they also suffer from largely unexplained pulmonary fibrosis and inflammatory lesions. This hyper-inflammatory response is probably multifactorial, but we speculate that the reduced polyP contributes to the tissue damage via unregulated complement activation. Indeed, it is possible that alterations in interactions between polyP and C1-INH may contribute to several inflammatory disorders.
In response to injury, polyP likely accumulates at very high concentrations on damaged cell surfaces where platelet-rich thrombi form, and where C1-INH also is found. Although in vivo concentrations of polyP at sites of injury have not been directly measured, based on the known concentration of polyP in platelets (>0.7 nmol/108 platelets)30 and the efficiency of release from dense granules during thrombin-induced activation (∼80%), concentrations of polyP could readily exceed 500 μM at sites of vascular injury and thrombus formation.35 Such levels of polyP would readily dampen complement activation by potentiating the inhibitory properties of C1-INH and interfering with the terminal pathway,38 overall protecting the underlying host cells from innate immune-mediated destruction. Recent reports that polyP with lengths of ≥70 monoP units induces inflammatory changes to endothelial cells63,64 may be explained by differential effects of polyP that are size dependent, as noted above, as well as the context. Thus, C1-INH and/or C5b,6 may act as switches, their presence favoring an anti-inflammatory role for polyP.
C1-INH exhibits a wide range of anti-inflammatory and vasculoprotective effects in vitro and in vivo.65,66 These are based primarily on its role as a serpin, but also through noncovalent interactions with multiple proteins. Whether these interactions are affected by polyanions, such as heparin/heparan sulfate or polyP is not known. However, C1-INH binds to extracellular matrix proteins,67 which may help localize it to sites of inflammation, interfere with formation of the alternative pathway C3 convertase, dampen leukocyte-endothelial interactions,68 and block gram-negative endotoxins and lipopolysaccharide.69 Plasma-derived and recombinant forms of C1-INH are widely used to treat hereditary angioedema,70,71 and in preclinical and limited clinical studies, have also shown benefit for gram-negative sepsis, pulmonary vascular leak syndromes, organ transplant rejection, and myocardial ischemia-reperfusion injury.65,66 The role of polyanions in modulating these properties of C1-INH, and thus its therapeutic efficacy beyond hereditary angioedema has not been examined, but may be clinically relevant. Our studies that reveal the existence of a naturally occurring polyanionic cofactor for C1-INH in the form of polyP, provide vital new insights into the physiological function of this important anti-inflammatory serpin.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank A. Johnson of the University of British Columbia Flow Facility for help in establishing flow cytometry protocols.
R.N.P. was supported by the National Health and Medical Research Council of Australia and the Australian Research Council. H.K. was supported by an operating grant and a Clinician-Scientist Award from the Canadian Institutes of Health Research. E.M.C. was supported by grants from the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada, and the Canada Foundations for Innovation. E.M.C. holds a CSL Behring Research Chair and a Tier 1 Canada Research Chair in Endothelial Cell Biology, and is an Adjunct Scientist with the Canadian Blood Services. Laboratory studies were supported by a grant from the National Institutes of Health National Heart, Lung, and Blood Institute (R01 HL047014) (J.H.M.). None of the funders had any input into the design of the experiments, the collection, analysis, or interpretation of the data, the writing of the manuscript, or the decision to submit it for publication.
Authorship
Contribution: L.C.W., E.L., R.N.P., and E.M.C. designed experiments, analyzed data, wrote the manuscript, and managed the project; L.C.W., E.L., L.H., R.C.D., P.R.K., T.S., R.J.T., V.L., S.A.S., H.K., and J.H.M. prepared reagents and performed experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Edward M. Conway, University of British Columbia Centre for Blood Research, 2350 Health Sciences Mall, #4303, Vancouver, BC V6T 1Z3, Canada; e-mail: ed.conway@ubc.ca; and Robert N. Pike, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, VIC 3086, Australia; e-mail: r.pike@latrobe.edu.au.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal