

To the editor:
Rituximab, a chimeric CD20 monoclonal antibody that causes depletion of B cells, shows short-term treatment efficacy in 40% to 60% of immune thrombocytopenia (ITP) patients.1,2 Our recently published multicenter randomized open-label phase 2 trial comparing 3 rituximab dosing schemes showed 50% responses in 138 ITP patients.3 Unfortunately, the treatment efficacy of rituximab so far cannot be predicted. Here, however, we show absent levels of platelet-bound antibodies to be associated with refractoriness to rituximab. Assays on platelet-bound antibodies may hence lead to a more personalized treatment approach for these patients.
ITP was diagnosed in accordance with the recommendation of the American Society of Hematology (ASH),4 by patient and family history, physical examination, and laboratory investigations (eg, normal white blood cell count and differentiation, normal red blood cell count, red blood cell indices and mean platelet volume, absence of HIV, hepatitis B virus, hepatitis C virus, Helicobacter pylori, and antinuclear factor, antinuclear antibody, antiphospholipid antibodies). Furthermore, low to normal thrombopoietin levels supported thrombocytopenia to be due to increased platelet destruction.5 Eligible patients were 18 years of age or older, with an ITP relapse or refractoriness (minimally 2 platelet counts <30 × 109/L), at least 3 weeks after high-dose (≥1 mg/kg) corticosteroids, who were randomized between 3 rituximab dosing schemes, that is, 2 or 4 once-weekly standard 375 mg/m2 doses and a twice-weekly 750 mg/m2 regimen.3 Complete response (CR), good/partial response (PR), and moderate response (MR) were defined as platelet counts of 150 × 109/L or more, 50 × 109/L or more on 2 consecutive occasions, and a platelet count over 30 × 109/L with at least twice the baseline count, respectively. The different dosages of rituximab did not lead to changes in response rate.3
Prior to starting the first rituximab dose and subsequently for 10 weeks, weekly EDTA-anticoagulated blood sampling was requested to assess the presence of platelet-associated immunoglobulin G (IgG) and IgM autoantibodies by the direct platelet immunofluorescence test (PIFT) and the eluate-(indirect) PIFT, as described previously.6 In the current report, a positive “PIFT” refers to the combination of a reactive (= 1+ to 3+) direct as well as eluate PIFT. One or both tests with negative reactions were defined as a negative “PIFT.” Additionally, in samples with sufficient platelet numbers, both the PIFT and a direct monoclonal antibody immobilization of platelet antigens (MAIPA) assay were performed; the latter to detect IgG-class autoantibodies.7 All monoclonal antibodies, CLBthromb/1 (CD41, anti-glycoprotein IIb [GPIIb]), MB45 (CD42a, anti-GPIX), and SW16 (CD42d, anti-GPV) were supplied by Sanquin Reagents (Amsterdam, The Netherlands).
For statistical analysis, correlation between continuous variables was calculated by the Spearman rank test, association between categorical valuables was calculated by the 2-tailed Fisher exact probability test, and 95% confidence intervals (CIs) were calculated using Stata version 14.0 (StataCorp LP).
Prerituximab treatment samples from 112 of 138 patients and follow-up samples during and after rituximab treatment from 80 patients were sent to our laboratory (supplemental Figure 1, available on the Blood Web site).
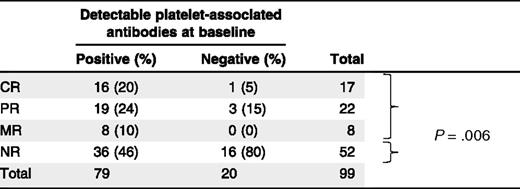
In 99 of 112 patient samples (88%), the platelet count in prerituximab treatment samples was sufficient to enable platelet-associated autoantibody detection by PIFT. Of these, a representative 47 (47%) responded to rituximab (CR, n = 17; PR, n = 22; and MR, n = 8, respectively) whereas 52 (53%) were nonresponders (NRs). For all tested patients (n = 99), direct PIFT results corresponded with the eluate-PIFT results. Antibodies were present in 79 patients, of whom 43 responded (54%) with 16 complete (21%). In contrast, the absence of antibodies in 20 patients was associated with 4 responses (20%) of which only 1 (5%) was complete (Table 1; P = .006). In summary, undetectable platelet autoantibodies in the direct PIFT strongly predict absence of or less than complete response to rituximab (Table 1; negative predictive value, 95.0%; CI, 73.2%-99.25%, positive predictive value, 20.3%; CI, 17.7%-23.1%).
Platelet-associated antibody detection prior to initiation with rituximab
 |
|
In the NR group, we found significantly (P = .006) more patients without detectable autoantibodies than in the response group (CR + PR + MR).
Vice versa, 16 patients (94%) with a complete response to rituximab showed positive to very-strong-positive direct PIFT reactions (mean level, 2+) whereas the percentage of nonresponsive patients with a positive direct PIFT was much lower (69%; P = .051). In patients with partial or moderate platelet responses, positive PIFT occurred with similar frequency in 19 (86%) and 8 (100%), respectively.
Serial autoantibody testing was performed for 80 of the sampled patients (71%). Of these, the presence of platelet autoantibodies and corresponding platelet counts were evaluated “per response group” at baseline and at the time of the highest platelet counts that were reached within 10 weeks after start of rituximab treatment (Figure 1; supplemental Figure 2). Of 30 patients predominantly responding (10 CR, 10 PR, 3 MR), the platelet count also enabled the direct MAIPA. Both PIFT and MAIPA results in these patients appeared in full concordance, that is, 7 patients (4 NR and 3 PR) were negative with both PIFT and direct MAIPA and 23 (3 NR, 3 MR, 7 PR, and 10 CR) positive with both PIFT and direct MAIPA.

Direct PIFT results and platelet counts prior to initiation with rituximab and at the time of the highest platelet counts reached within 10 weeks after start of rituximab treatment of 80 serially tested ITP patients. X-axis for panel B also applies for panel A. (A) CR, n = 16; PR, n = 16; MR, n = 8; NR, n = 40. Direct PIFT scores before (pre) and at the time of the highest platelet count within 10 weeks after (post) treatment with rituximab: 0, negative; 1, positive; 2, strong positive; and 3, very strong positive. Changes in antibody detection results are indicated by means of connecting lines. (B) Platelet counts before and the highest platelet count within 10 weeks after treatment with rituximab.
Direct PIFT results and platelet counts prior to initiation with rituximab and at the time of the highest platelet counts reached within 10 weeks after start of rituximab treatment of 80 serially tested ITP patients. X-axis for panel B also applies for panel A. (A) CR, n = 16; PR, n = 16; MR, n = 8; NR, n = 40. Direct PIFT scores before (pre) and at the time of the highest platelet count within 10 weeks after (post) treatment with rituximab: 0, negative; 1, positive; 2, strong positive; and 3, very strong positive. Changes in antibody detection results are indicated by means of connecting lines. (B) Platelet counts before and the highest platelet count within 10 weeks after treatment with rituximab.
CR in the Haemato Oncology Foundation for Adults in The Netherlands (HOVON) 64 Study was defined as a rise in platelet counts to 150 × 109/L or more. Nowadays, ITP is defined as platelet counts <100 × 109/L.8 Recategorization of the response groups, using 100 × 109/L did not result in different response numbers.
Anti-CD20 monoclonal antibodies deplete B cells for periods varying from months to >1 year. In this respect, B cells are essential to present antigens to CD4+ T cells and moreover secrete cytokines-activating macrophages, dendritic cells, and immune-regulatory cells and thus an ongoing autoimmune response.9,-11 Additionally, rituximab is reported to normalize both the abnormalities and dysfunction of the T-cell compartment in ITP patients.9,12,-14 However, depletion of pre–plasma cell B cells, and with it decreased autoantibody production, is regarded as the most likely mechanism of action. But, so far not one of these explanations could be correlated to rituximab’s variable therapeutic effects.
Our results are in agreement with the latter mechanism, whereas all serially tested CR patients (n = 16) showed decreasing antibody results and 32 of 39 nonresponsive patients have nondecreasing autoantibodies after or undetectable autoantibodies before rituximab treatment (Figure 1). A possible explanation for nondecreasing antibodies could be insufficient eradication of antibody-producing plasma cells. Indeed, long-lived plasma cells were detected in the spleen of ITP patients nonresponsive to rituximab treatment and it has been suggested that B-cell depletion promotes the differentiation and settlement of these long-lived plasma cells in the spleen.15 Although nonresponsive patients without detectable platelet-associated autoantibodies fit the ASH criteria for ITP, these patients might represent a subgroup of ITP patients with possibly T-cell–mediated platelet destruction. Indeed, Audia et al in this respect showed the importance of activated splenic CD8+ T cells in ITP patients unresponsive to rituximab.16 Furthermore, CD8+ T cells were shown to contribute to the murine splenocyte’s ability to induce thrombocytopenia; recently, Arthur et al showed CD8+ T-cell–mediated antibody-independent platelet clearance in a murine model for refractoriness to platelet transfusions.17,18
Immune-suppressive treatment in combination with rituximab, as recently published, may improve the limited response for rituximab as single treatment.19 In this respect, it will be interesting to study the effect of combination therapy in ITP patients with and without detectable platelet autoantibodies.
In conclusion, our results show absence of platelet-bound antibodies to be associated with a low response to rituximab. Additionally, response to rituximab appeared strongly associated with a decline in platelet-bound antibodies. Our findings importantly implicate that both antibody negativity vs strong antibody presence might enable a more individualized therapeutic approach in this group of ITP patients.
The online version of this article contains a data supplement.
Authorship
Contribution: L.P. was principal investigator, carried out the initial analysis, reviewed and revised the manuscript, and approved the final manuscript as submitted; E.H. designed the data collection instrument, coordinated and supervised data collection, critically reviewed the manuscript, and approved the final manuscript as submitted; M.S. submitted samples and helped analyze the initial rituximab data; B.v.d.H. coordinated and supervised data collection; L.P., J.J.Z., and M.d.H. conceptualized and designed the study, drafted the initial manuscript, and approved the final manuscript as submitted; and the HOVON 64 Study Group included patients and executed the trial.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the Dutch HOVON 64 Study Group appears in “Appendix.”
Correspondence: Leendert Porcelijn, Immunohematology Diagnostics, Plesmanlaan 125 (Q2), 1066CX Amsterdam, The Netherlands; e-mail: l.porcelijn@sanquin.nl.
Appendix: study group members
The members of the Dutch HOVON 64 Study Group are: Jaap J. Zwaginga (Department of Immunohematology and Blood Transfusion, Leiden University Medical Centre and the Centre for Clinical Transfusion Research, Sanquin-Leiden University Medical Centre, Leiden, The Netherlands), Bronno van der Holt (HOVON Data Center, Erasmus MC Cancer Institute–Clinical Trial Centre, Rotterdam, The Netherlands), Peter A. te Boekhorst (Department of Hematology, Erasmus MC, Rotterdam, The Netherlands), Bart J. Biemond (Department of Hematology, Academic Medical Centre Amsterdam, Amsterdam, The Netherlands), Mark-David Levin (Department of Internal Medicine, Albert Schweitzer Hospital, Dordrecht, The Netherlands), René van der Griend (Department of Internal Medicine, Diakonessenhuis, Utrecht, The Netherlands), Anneke Brand (Department of Immunohematology and Blood Transfusion, Leiden University Medical Centre and the Centre for Clinical Transfusion Research, Sanquin-Leiden University Medical Centre, Leiden, The Netherlands), Sonja Zweegman (Department of Hematology, VU University Medical Centre, Amsterdam, The Netherlands), Hans F. M. Pruijt (Department of Internal Medicine, Jeroen Bosch Hospital, Den Bosch, The Netherlands), Vera M. J. Novotny (Department of Hematology, Radboud University Medical Centre, Nijmegen, The Netherlands), Art Vreugdenhil (Department of Internal Medicine, Máxima Medical Centre, Veldhoven, The Netherlands), Marco R. de Groot (Department of Internal Medicine, Medisch Spectrum Twente, Enschede, The Netherlands), Okke de Weerdt (Department of Internal Medicine, St. Antonius Hospital, Nieuwegein, The Netherlands), Elisabeth C. M. van Pampus (Department of Laboratory Medicine, Radboud University Medical Centre, Nijmegen, The Netherlands), Tanja M. van Maanen-Lamme (Department of Internal Medicine, Westfriesgasthuis, Hoorn, The Netherlands), Shulamiet Wittebol (Department of Internal Medicine, Meander Hospital, Amersfoort, The Netherlands), Martin R. Schipperus (Department of Internal Medicine, HagaZiekenhuis, Den Haag, The Netherlands), Matthijs H. Silbermann (Department of Internal Medicine, Tergooiziekenhuizen, Blaricum, The Netherlands), Peter C. Huijgens (Department of Hematology, VU University Medical Centre, Amsterdam, The Netherlands), Marleen Luten (HOVON Data Center, Erasmus MC Cancer Institute–Clinical Trial Centre, Rotterdam, The Netherlands), Rene Hollestein (HOVON Data Center, Erasmus MC Cancer Institute–Clinical Trial Centre, Rotterdam, The Netherlands), Jan A. C. Brakenhoff (Department of Internal Medicine, Waterland Hospital, Purmerend, The Netherlands), Jolanda G. Schrama (Department of Internal Medicine, Spaarne Hospital, Hoofddorp, The Netherlands), Fransje A. A. Valster (Department of Internal Medicine, Lievensberg Hospital, Bergen op Zoom, The Netherlands), Gerjo A. Velders (Department of Internal Medicine, Gelderse Vallei Hospital, Ede, The Netherlands), and Harry R. Koene (Department of Internal Medicine, St. Antonius Hospital, Nieuwegein, The Netherlands).

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal