

Key Points
Degree of TAFI activation in HA is a modifier of hemophilic joint bleeding that inversely affects bleeding severity.
Defective TAFI activation in severe congenital HA impairs protection against uPA-mediated fibrinolysis in bleeding joints.

Abstract
Joint bleeds are common in congenital hemophilia but rare in acquired hemophilia A (aHA) for reasons unknown. To identify key mechanisms responsible for joint-specific bleeding in congenital hemophilia, bleeding phenotypes after joint injury and tail transection were compared in aHA wild-type (WT) mice (receiving an anti–factor VIII [FVIII] antibody) and congenital HA (FVIII−/−) mice. Both aHA and FVIII−/− mice bled severely after tail transection, but consistent with clinical findings, joint bleeding was notably milder in aHA compared with FVIII−/− mice. Focus was directed to thrombin-activatable fibrinolysis inhibitor (TAFI) to determine its potentially protective effect on joint bleeding in aHA. Joint bleeding in TAFI−/− mice with anti-FVIII antibody was increased, compared with WT aHA mice, and became indistinguishable from joint bleeding in FVIII−/− mice. Measurements of circulating TAFI zymogen consumption after joint injury indicated severely defective TAFI activation in FVIII−/− mice in vivo, consistent with previous in vitro analyses in FVIII-deficient plasma. In contrast, notable TAFI activation was observed in aHA mice, suggesting that TAFI protected aHA joints against bleeding. Pharmacological inhibitors of fibrinolysis revealed that urokinase-type plasminogen activator (uPA)–induced fibrinolysis drove joint bleeding, whereas tissue-type plasminogen activator–mediated fibrinolysis contributed to tail bleeding. These data identify TAFI as an important modifier of hemophilic joint bleeding in aHA by inhibiting uPA-mediated fibrinolysis. Moreover, our data suggest that bleed protection by TAFI was absent in congenital FVIII−/− mice because of severely defective TAFI activation, underscoring the importance of clot protection in addition to clot formation when considering prohemostatic strategies for hemophilic joint bleeding.
Introduction
Patients with severe factor VIII (FVIII) or FIX deficiency (hemophilia A [HA] or B) experience frequent joint bleeding that results in hemophilic joint disease, a major cause of joint deterioration and disability in the hemophilia population.1 Joint bleeds account for ∼70% of total bleeds in patients with severe hemophilia (factor levels <1%), with the incidence of joint bleeds generally increasing with the severity of FVIII or FIX deficiency. Factor replacement therapy limits the incidence of joint bleeds to a certain degree but cannot prevent the progression of hemophilic joint disease, even when prophylaxis is initiated very early in life.2-4 Furthermore, the development of inhibitory alloantibodies in ∼30% of patients with hemophilia complicates lifelong factor replacement therapy and increases susceptibility to frequent joint bleeds.5
Unlike in congenital hemophilia, joint bleeding is uncommon in acquired HA (aHA), a rare autoimmune disease, in which autoantibodies are produced against certain epitopes on endogenous FVIII, and these type 2 inhibitors typically do not accomplish full inhibition of FVIII.6 This divergent clinical bleeding pattern provides unique opportunities to interrogate the key mechanism(s) responsible for joint-specific bleeding in congenital HA. Such mechanisms may extend well beyond the initial clot formation and involve factors that modulate vascular bed–specific clot stability and/or fibrinolysis.
One potential candidate for modulation of clot stability and fibrinolysis is thrombin-activatable fibrinolysis inhibitor (TAFI; gene CPB2).7,8 Activated TAFI (TAFIa) inhibits fibrinolysis by removing C-terminal Lys residues on partially degraded fibrin, which serve as a cofactor for plasminogen activation by tissue-type plasminogen activator (tPA) or urokinase-type plasminogen activator (uPA).9-11 It is generally recognized that bleeding is precipitated in hemophilia by defective thrombin generation in tissues with relatively low tissue factor expression, such as the joints.12 Hemophilic clots in these tissues are fragile and susceptible to premature degradation by the fibrinolytic system.13 At least in vitro, defective TAFI activation in hemophilia has been shown to enhance fibrinolysis, resulting in poor clot stability and premature lysis.14-16 Relatively abundant thrombin is required for TAFI activation that is normally generated by thrombin-mediated FXI activation and feedback via the intrinsic pathway.17 TAFI activation is defective in hemophilia plasma in vitro because this intrinsic feedback loop for thrombin generation is inoperable in hemophilia, especially at low tissue factor levels. However, the activation of TAFI and TAFIa-mediated inhibition of fibrinolysis are relatively unaffected in the presence of an inhibitory anti-FVIII antibody in normal plasma, even at antibody concentrations that severely affect clotting.18 Therefore, the susceptibility of TAFI activation to residual levels of FVIII provides a notable distinction between acquired and congenital hemophilia in clot lysis assays in vitro.
Our understanding of the physiological and pathophysiological activation mechanisms of TAFI in various vascular beds in vivo remains limited, and the extent to which the activation of TAFI in hemophilia in vivo is defective remains undetermined. This determination is convoluted by the fact that thrombomodulin enhances thrombin-mediated TAFI activation by approximately 1000-fold.19,20 Consequently, TAFI activation in the presence of thrombomodulin proceeds efficiently in hemophilia plasma even at low tissue factor concentrations.21 Thrombomodulin is readily detected on the synovial lining of the joint.22,23 The degree of TAFI activation in hemophilic joints is therefore conceivably dependent on the vascular bed–specific contribution of thrombomodulin.
To elucidate the potential contribution of TAFI in conjunction with vascular bed–specific factors to hemophilic joint bleeding, we adopted a joint injury model in FVIII−/− mice to induce joint bleeding.24 The joint injury model is distinctive from other frequently used acute bleeding models in hemophilia (eg, tail clip or saphenous vein puncture25 ) in that bleeding occurs slowly over several days into the microvascularized intra- and periarticular tissues. Comparing bleeding characteristics of the joint bleeding model with those of an acute bleeding model, such as tail transection, may help to discern time-dependent and vascular bed–specific contributions to the complex mechanisms of hemophilic joint and soft tissue bleeding.
Here, we aimed to identify key factors involved in joint-specific bleeding in HA by comparing the bleeding phenotype of acquired vs congenital FVIII deficiency in mice. We hypothesized that impaired generation of TAFIa in severe congenital hemophilia underlies joint bleeding and, conversely, that residual TAFI activation occurring in acquired hemophilia protects against joint bleeding.
Materials and methods
Additional details can be found in the supplemental Information, available on the Blood Web site.
Animals
All animal protocols were approved by the institutional animal and care committee of The Scripps Research Institute. FVIII−/− mice on a BALB/c background26 were a generous gift from Dr David Lillicrap (Queen’s University, Kingston, Ontario, Canada). TAFI−/− mice on a C57Bl/6J background were generated as described.27 Wild-type (WT) BALB/c and WT C57Bl/6J mice were obtained from the Scripps Research Institute internal breeding facility. Skeletally mature mice (age 12-16 weeks) of both sexes were used for joint bleeding assays, and mice (age 8-12 weeks) of both sexes were used for tail bleeding assays.
Joint and tail bleeding assays
Determination of TAFI activation in plasma
TAFI activation in vivo was determined by the extent of TAFI zymogen consumption. TAFI zymogen levels were determined in mouse plasma samples using immunoprecipitation and immunoblot analyses.31
Treatments
Two hours before joint injury or tail resection, mice were injected retroorbitally with inhibitory anti-FVIII antibody GMA-801518,32 (0.25-1 mg/kg; Green Mountain Antibodies, Burlington, VT), inhibitory anti-TAFI antibody MA-RT36A3F533 (destabilizing TAFIa; 7.5 mg/kg) or MA-TCK26D634,35 (inhibiting thrombin- and plasmin-mediated activation of TAFI and also interfering with TAFIa binding to fibrin; 3.75 mg/kg), or an inhibitory anti-uPA antibody (mU1; 10 mg/kg)36 diluted in sterile saline (0.9%; Hospira Inc.). Tranexamic acid (TXA; Acros Organics) was administered in the drinking water as described.37 uPA inhibitor amiloride (Sigma Aldrich) was administered subcutaneously using an osmotic pump (Alzet #2001) at a rate of 4 mg/kg per day.
Statistical analysis
Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test for in vivo data or 1-way analysis of variance followed by Dunnett’s post hoc multiple comparison test for thrombin generation and clot lysis data were used to determine statistical significance using GraphPad Prism (San Diego, CA). Survival curves were graphed using the Kaplan-Meier method, and statistical significance was determined by the log-rank (Mantel-Cox) test. Correlations were analyzed using the Pearson correlation test. A P value of <.05 was considered statistically significant. Data are expressed as mean ± standard deviation.
Results
Differences in joint bleeding susceptibility between aHA vs congenital HA mouse models
Joint injury in FVIII−/− BALB/c mice caused considerable joint bleeding, as evidenced by a decrease in Hct 2 days after injury (day 2 [D2] Hct, 29% ± 11% vs baseline Hct, 47% ± 2%; P < .0001; Figure 1A), and was consistent with previous studies.24,38 Inhibitory anti-FVIII antibody GMA-8015 was used to induce aHA. A GMA-8015 dose of 0.25 mg/kg in WT BALB/c mice, corresponding to 35 BU/mL at 2 hours after injection, was previously shown to increase tail bleeding to the same extent as in FVIII−/− BALB/c mice.18 Also, similar tail bleeding was observed when the inhibitory anti-FVIII antibody was administered to WT C57Bl/6J mice (Figure 2A-C), indicating that there were no apparent differences between C57Bl/6J and BALB/c strains, although FVIII−/− C57BL/6J mice were not tested. However, only minimal joint bleeding was observed in anti-FVIII antibody–treated WT C57Bl/6J mice (D2 Hct, 40% ± 4%; Figure 1A) or anti-FVIII antibody–treated WT BALB/c mice (D2 Hct, 44% ± 4%; supplemental Figure 1). Increasing the anti-FVIII antibody dosage to 1 mg/kg in C57Bl/6J mice resulted in significant joint bleeding (D2 Hct, 36% ± 9%; P < .05); however, bleeding remained considerably less severe than observed in FVIII−/− mice (Figure 1A). Weight-bearing impairment of the injured hind leg was measured as a functional consequence of joint bleeding based on its inverse correlation with D2 Hct (P < .0001; r = −0.80; supplemental Figure 2). Consistent with less severe bleeding, anti-FVIII antibody–treated WT mice showed impaired but nonsignificant imbalanced weight bearing compared with baseline(Figure 1B). In contrast, the weight-bearing ratio was significantly impaired in injured FVIII−/− mice (Figure 1B). Thus, the joint injury model recapitulates the clinically observed discrepancy in joint bleeding susceptibility between patients with congenital HA and those with aHA.
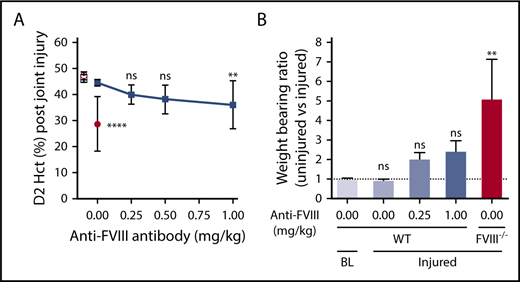
aHA mice are protected against joint bleeding after injury. (A) Joint bleeding after knee injury in WT C57Bl/6J mice (n = 9-12; filled squares) that received different doses of the inhibitory anti-FVIII antibody (0-1 mg/kg; GMA-8015). Bleeding was determined by measuring Hct at day 2 (D2) postinjury. D2 Hct values for injured FVIII−/− BALB/c mice (n = 9; filled circles) are shown to illustrate typical joint bleeding in congenital HA mice. Hct of WT C57Bl/6J (open square) or FVIII−/− BALB/c (open circle) mice at baseline (BL; no injury) are provided as a no-bleeding reference (n = 8-10). (B) Weight-bearing ratio of left (uninjured) vs right (injured) hind paw at BL (n = 5) and at D2 after right knee injury in WT C57Bl/6J (n = 6) or FVIII−/− BALB/c (n = 4) mice at the indicated doses (mg/kg) of anti-FVIII antibody. Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test was used to compare experimental mice with WT mice at baseline. BL indicates baseline without knee injury; injured indicates right knee injury. **P < .01, ****P < .0001. ns, not significant.
aHA mice are protected against joint bleeding after injury. (A) Joint bleeding after knee injury in WT C57Bl/6J mice (n = 9-12; filled squares) that received different doses of the inhibitory anti-FVIII antibody (0-1 mg/kg; GMA-8015). Bleeding was determined by measuring Hct at day 2 (D2) postinjury. D2 Hct values for injured FVIII−/− BALB/c mice (n = 9; filled circles) are shown to illustrate typical joint bleeding in congenital HA mice. Hct of WT C57Bl/6J (open square) or FVIII−/− BALB/c (open circle) mice at baseline (BL; no injury) are provided as a no-bleeding reference (n = 8-10). (B) Weight-bearing ratio of left (uninjured) vs right (injured) hind paw at BL (n = 5) and at D2 after right knee injury in WT C57Bl/6J (n = 6) or FVIII−/− BALB/c (n = 4) mice at the indicated doses (mg/kg) of anti-FVIII antibody. Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test was used to compare experimental mice with WT mice at baseline. BL indicates baseline without knee injury; injured indicates right knee injury. **P < .01, ****P < .0001. ns, not significant.
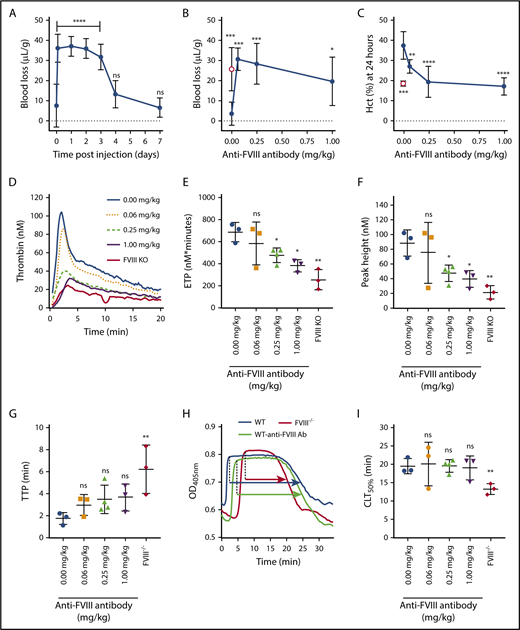
aHA mice are not protected against tail bleeding. (A) Tail bleeding in WT C57Bl/6J mice at different time points after injection of anti-FVIII antibody (0.25 mg/kg; n = 4-5). Bleeding was determined by measuring the mouse weight-normalized blood loss (µL/g) after tail resection for 20 minutes. (B) Tail bleeding in WT C57Bl/6J mice (n = 5-10; filled circles) measured 2 hours after injection of different doses of anti-FVIII antibody (0-1 mg/kg). Tail bleeding in FVIII−/− BALB/c mice (n = 8; open circle) is shown as reference. (C) Bleeding determined by Hct at 24 hours after tail resection in WT C57Bl/6J mice (n = 5-10; filled circles) injected with different doses of anti-FVIII antibody (0-1 mg/kg). Hct at 24 hours in FVIII−/− BALB/c mice (n = 8; open circle) after tail resection is shown as reference. Representative thrombin-generation profiles (D), endogenous thrombin potential (ETP; nM × minutes) (E), peak height (nM) (F), and time to peak (TTP; minutes) (G) of plasma samples taken 24 hours after injection of anti-FVIII antibody (0-1 mg/kg) in WT BALB/c mice (n = 3-4 per antibody dosage) or FVIII−/− BALB/c mice (n = 3). (H) Representative uPA-mediated plasma clot lysis profiles from a FVIII−/− BALB/c mouse (red) or WT BALB/c mouse injected 24 hours before with either saline (blue) or anti-FVIII antibody (0.25 mg/kg; green). The 50% clot lysis time (CLT50%), representing the time between maximal clot formation and half-maximal clot lysis, is indicated by arrows. (I) CLT50% (minutes) of plasma samples taken 24 hours after injection of anti-FVIII antibody (0-1 mg/kg) in WT BALB/c mice (n = 3-4 per antibody dosage) or FVIII−/− BALB/c mice (n = 3). Thrombin-generation and clot lysis data are the mean of 2 independent experiments for each sample. Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test (A-C) or 1-way analysis of variance followed by Dunnett’s post hoc multiple comparison test (D-I) was used to compare experimental mice with WT mice injected with saline. *P < .05, **P < .01, ***P < .001, ****P < .0001. KO, knockout; ns, not significant; OD, optical density.
aHA mice are not protected against tail bleeding. (A) Tail bleeding in WT C57Bl/6J mice at different time points after injection of anti-FVIII antibody (0.25 mg/kg; n = 4-5). Bleeding was determined by measuring the mouse weight-normalized blood loss (µL/g) after tail resection for 20 minutes. (B) Tail bleeding in WT C57Bl/6J mice (n = 5-10; filled circles) measured 2 hours after injection of different doses of anti-FVIII antibody (0-1 mg/kg). Tail bleeding in FVIII−/− BALB/c mice (n = 8; open circle) is shown as reference. (C) Bleeding determined by Hct at 24 hours after tail resection in WT C57Bl/6J mice (n = 5-10; filled circles) injected with different doses of anti-FVIII antibody (0-1 mg/kg). Hct at 24 hours in FVIII−/− BALB/c mice (n = 8; open circle) after tail resection is shown as reference. Representative thrombin-generation profiles (D), endogenous thrombin potential (ETP; nM × minutes) (E), peak height (nM) (F), and time to peak (TTP; minutes) (G) of plasma samples taken 24 hours after injection of anti-FVIII antibody (0-1 mg/kg) in WT BALB/c mice (n = 3-4 per antibody dosage) or FVIII−/− BALB/c mice (n = 3). (H) Representative uPA-mediated plasma clot lysis profiles from a FVIII−/− BALB/c mouse (red) or WT BALB/c mouse injected 24 hours before with either saline (blue) or anti-FVIII antibody (0.25 mg/kg; green). The 50% clot lysis time (CLT50%), representing the time between maximal clot formation and half-maximal clot lysis, is indicated by arrows. (I) CLT50% (minutes) of plasma samples taken 24 hours after injection of anti-FVIII antibody (0-1 mg/kg) in WT BALB/c mice (n = 3-4 per antibody dosage) or FVIII−/− BALB/c mice (n = 3). Thrombin-generation and clot lysis data are the mean of 2 independent experiments for each sample. Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test (A-C) or 1-way analysis of variance followed by Dunnett’s post hoc multiple comparison test (D-I) was used to compare experimental mice with WT mice injected with saline. *P < .05, **P < .01, ***P < .001, ****P < .0001. KO, knockout; ns, not significant; OD, optical density.
No susceptibility differences in tail bleeding between aHA and congenital HA mouse models
Unlike with reduced susceptibility to joint bleeding, GMA-8015 anti-FVIII antibody–treated WT mice demonstrated no susceptibility difference compared with FVIII−/− mice in acute tail bleeding. Blood loss volumes in anti-FVIII antibody–treated WT C57Bl/6J (Figure 2A) and WT BALB/c18 mice were similar to those in FVIII−/− BALB/c mice after tail resection (Figure 2B). Several controls were performed to ensure that experimental factors (eg, differences in the timeframe and analysis methods for bleeding) were not responsible for the observed difference in the susceptibility to joint and tail bleeding. First, the clearance rate of GMA-8015 was determined. When administered at 0.25 mg/kg, the antibody increased tail bleeding up to 3 days after injection to levels similar to those observed in FVIII−/− mice (Figure 2A), thus ruling out that loss of FVIII inhibition could be responsible for reduced joint bleeding in aHA mice. Second, a GMA-8015 dose-response titration demonstrated that FVIII−/−like tail bleeding was induced from ≥0.0625 mg/kg of GMA-8015, indicating that the 0.25-mg/kg dose was well within the GMA-8015 dosing range that caused significant inhibition of coagulation. Finally, Hct was determined after tail resection to compare tail bleeding with joint bleeding using the same end point measurement. Hct at 24 hours was found to be inversely correlated with acute blood loss after 20 minutes (P < .0001; r = −0.73; supplemental Figure 3). For example, GMA-8015 at 0.25 mg/kg, which caused similar acute blood loss after tail resection in WT compared with FVIII−/− mice (Figure 2B), also reduced Hct at 24 hours to a similar extent (20% ± 8% vs 19% ± 1%; Figure 2C). At 0.0625 mg/kg, however, acute blood loss was still maximal, whereas Hct at 24 hours was intermediate (27% ± 3% vs 37% ± 7%), indicating fewer rebleeding events. This dosage-dependent effect at 24 hours could not be explained by the ex vivo APTT that was maximally prolonged at 0.0625 mg/kg (supplemental Figure 4A). In contrast, dose-dependent differences did arise in ex vivo thrombin generation, with 0.25 mg/kg of antibody having a significant impact (reduced endogenous thrombin potential and peak height), whereas 0.0625 mg/kg did not (Figure 2D-G). Thus, GMA-8015 at a dosage of 0.25 mg/kg provided a prolonged impairment of coagulation sufficient to increase tail bleeding maximally, but not joint bleeding. Unlike in FVIII−/− mouse plasma, injection of GMA-8015 (≤1 mg/kg) did not impair the TAFIa-mediated attenuation of clot lysis in WT plasma (Figure 2H-I; supplemental Figure 4B-C), suggesting that residual thrombin formation in aHA mice leads to sufficient activation of TAFI and results in a prohemostatic effect in the joints after injury.
TAFI protects against joint bleeding in aHA
To probe a potential role for TAFI in joint bleeding, the aHA model was applied to TAFI−/− mice. Administration of anti-FVIII antibody GMA-8015 (0.25 mg/kg) in TAFI−/− mice resulted in significant joint bleeding that was markedly increased compared with that in anti-FVIII antibody–treated WT mice (D2 Hct, 25% ± 8% vs 41% ± 3%; P < .0001; Figure 3A). Visual inspection indicated that hematomas in anti-FVIII antibody–treated TAFI−/− mice were similar to those in FVIII−/− mice and were larger and darker (blood filled) compared with those in anti-FVIII antibody–treated WT mice (Figure 3B). Increased joint bleeding in WT mice when the anti-FVIII antibody was coadministered with an inhibitory antibody against TAFI (MA-RT36A3F533 ) further confirmed that joint bleeding increased upon impaired TAFIa function (D2 Hct, 34% ± 7%; P < .01; Figure 3A). Similar results were obtained when the inhibitory anti-FVIII and anti-TAFI antibodies were coadministered in WT BALB/c mice, indicating that there were no apparent differences between C57Bl/6J and BALB/c strains (supplemental Figure 5). Histological analysis demonstrated presence of extravasated erythrocytes and local inflammation-induced hypercellularity in the joint space of FVIII−/− mice 7 days after injury (Figure 3C). This was associated with the expansion of the synovial and stromal linings (Figure 3D). Extravasation of erythrocytes was minimal in anti-FVIII antibody–treated WT mice, and although at day 4, some soft tissue expansion was observed in the joints of these mice, the extent of tissue expansion normalized at day 7 (Figure 3C-D). Coinciding with exposure to increased blood volumes, soft tissue expansion was overall increased in WT mice receiving both anti-FVIII and anti-TAFI antibodies and in anti-FVIII antibody–treated TAFI−/− mice (Figure 3C-D). Furthermore, the anti-TAFI antibody did not have an additional effect on joint bleeding or soft tissue expansion in FVIII−/− mice (supplemental Figure 6), suggesting that TAFI is indeed not being activated in hemophilia. Collectively, these data indicate that residual TAFI activation in aHA protects against extensive joint bleeding.
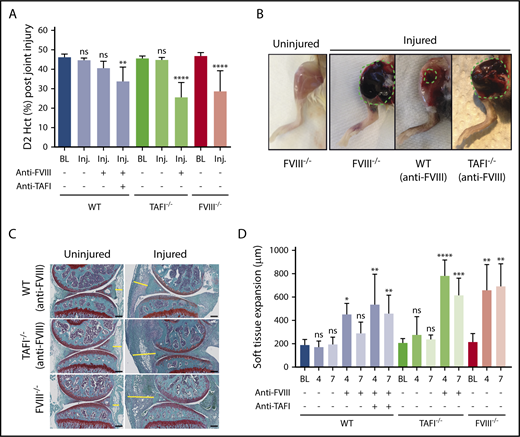
TAFI deficiency eliminates protection against joint bleeding in aHA mice. (A) Joint bleeding after knee injury in FVIII−/− BALB/c, WT C57Bl/6J, or TAFI−/− C57Bl/6J mice with or without anti-FVIII antibody (0.25 mg/kg) alone or in combination with anti-TAFI antibody MA-RT36A3F5 (7.5 mg/kg; n = 10-16). Bleeding was determined by measuring Hct at day 2 (D2) postinjury. (B) Photographs of the hematoma formed in the right injured knee at day 4 after injury in a FVIII−/− BALB/c mouse or an anti-FVIII antibody–treated WT-C57Bl/6J or TAFI−/− C57Bl/6J mouse. (C) Representative images of Safranin-O fast green staining of the left uninjured knee joint and corresponding right injured knee joint harvested at day 7 postinjury of a WT C57Bl/6J or TAFI−/− C57Bl/6J mouse injected with anti-FVIII antibody (0.25 mg/kg) or of a FVIII−/− BALB/c mouse. The yellow line indicates soft tissue expansion between the anterior meniscus and joint capsule. Original magnification, ×4; scale bar = 200 µm. (D) Soft tissue expansion (µm) at baseline (BL) and days 4 and 7 postinjury in WT C57Bl/6J, TAFI−/− C57Bl/6J, and FVIII−/− BALB/c mice with and without anti-FVIII antibody (0.25 mg/kg) and/or anti-TAFI antibody (MA-RT36A3F5; 7.5 mg/kg; n = 4-6). Soft tissue expansion was measured as the distance between the anterior meniscus and joint capsule (indicated by the yellow line in Figure 3C). BL indicates baseline without knee injury. Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test was used to compare experimental mice with their respective baseline controls. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant.
TAFI deficiency eliminates protection against joint bleeding in aHA mice. (A) Joint bleeding after knee injury in FVIII−/− BALB/c, WT C57Bl/6J, or TAFI−/− C57Bl/6J mice with or without anti-FVIII antibody (0.25 mg/kg) alone or in combination with anti-TAFI antibody MA-RT36A3F5 (7.5 mg/kg; n = 10-16). Bleeding was determined by measuring Hct at day 2 (D2) postinjury. (B) Photographs of the hematoma formed in the right injured knee at day 4 after injury in a FVIII−/− BALB/c mouse or an anti-FVIII antibody–treated WT-C57Bl/6J or TAFI−/− C57Bl/6J mouse. (C) Representative images of Safranin-O fast green staining of the left uninjured knee joint and corresponding right injured knee joint harvested at day 7 postinjury of a WT C57Bl/6J or TAFI−/− C57Bl/6J mouse injected with anti-FVIII antibody (0.25 mg/kg) or of a FVIII−/− BALB/c mouse. The yellow line indicates soft tissue expansion between the anterior meniscus and joint capsule. Original magnification, ×4; scale bar = 200 µm. (D) Soft tissue expansion (µm) at baseline (BL) and days 4 and 7 postinjury in WT C57Bl/6J, TAFI−/− C57Bl/6J, and FVIII−/− BALB/c mice with and without anti-FVIII antibody (0.25 mg/kg) and/or anti-TAFI antibody (MA-RT36A3F5; 7.5 mg/kg; n = 4-6). Soft tissue expansion was measured as the distance between the anterior meniscus and joint capsule (indicated by the yellow line in Figure 3C). BL indicates baseline without knee injury. Kruskal-Wallis test followed by Dunn’s post hoc multiple comparison test was used to compare experimental mice with their respective baseline controls. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant.
Defective TAFI activation in HA mice in vivo
The ability of TAFI to protect against extensive joint bleeding in aHA raised the question of whether the pronounced joint bleeding observed in the congenital HA mouse injury model resulted from defective TAFI activation. The extent of TAFI activation after knee injury was determined in plasma obtained from injured aHA WT mice and FVIII−/− mice by measuring zymogen consumption, as compared with the respective baseline levels. After injury, TAFI activation (inferred from the disappearance of TAFI zymogen) was detectable in anti-FVIII antibody–treated WT mice but not in FVIII−/− mice (Figure 4A-B). Treatment of FVIII−/− mice with recombinant human FVIII resulted in very low plasma zymogen TAFI levels after joint injury, indicating that the TAFI zymogen was almost completely consumed after FVIII administration. These in vivo data confirm important previous assumptions derived from in vitro studies,14,15 namely that TAFI activation in vivo is severely impaired upon joint injury with congenital FVIII deficiency. In contrast, notable TAFI activation remains detectable in acquired hemophilia.
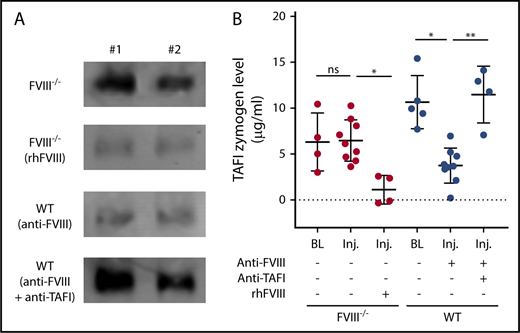
Defective TAFI activation after knee joint injury in FVIII−/− mice. (A) Representative examples (n = 2) of immunoblot signals (originating from the same blot with equal exposure) of zymogen TAFI levels precipitated from plasma samples 2 days after injury of FVIII−/− BALB/c without or with administration of recombinant human FVIII (rhFVIII; 200 IU/kg) or WT C57Bl/6J mice with anti-FVIII antibody (GMA-8015; 0.25 mg/kg) alone or in combination with anti-TAFI antibody (MA-TCK26D6; 3.75 mg/kg). (B) Quantification of plasma zymogen TAFI levels 2 days after injury by immunoblot after pull down in FVIII−/− mice without (n = 9) or with (n = 4) administration of rhFVIII (200 IU/kg) or in WT mice with anti-FVIII antibody (GMA-8015; 0.25 mg/kg) alone (n = 9) or in combination with anti-TAFI antibody (MA-TCK26D6; 3.75 mg/kg; n = 4). Kruskal-Wallis test was used followed by Dunn’s post hoc multiple comparison test to compare experimental mice. *P < .05, **P < .01. ns, not significant.
Defective TAFI activation after knee joint injury in FVIII−/− mice. (A) Representative examples (n = 2) of immunoblot signals (originating from the same blot with equal exposure) of zymogen TAFI levels precipitated from plasma samples 2 days after injury of FVIII−/− BALB/c without or with administration of recombinant human FVIII (rhFVIII; 200 IU/kg) or WT C57Bl/6J mice with anti-FVIII antibody (GMA-8015; 0.25 mg/kg) alone or in combination with anti-TAFI antibody (MA-TCK26D6; 3.75 mg/kg). (B) Quantification of plasma zymogen TAFI levels 2 days after injury by immunoblot after pull down in FVIII−/− mice without (n = 9) or with (n = 4) administration of rhFVIII (200 IU/kg) or in WT mice with anti-FVIII antibody (GMA-8015; 0.25 mg/kg) alone (n = 9) or in combination with anti-TAFI antibody (MA-TCK26D6; 3.75 mg/kg; n = 4). Kruskal-Wallis test was used followed by Dunn’s post hoc multiple comparison test to compare experimental mice. *P < .05, **P < .01. ns, not significant.
To determine the mechanism of TAFI activation in aHA WT mice after injury, the inhibitory anti-TAFI antibody MA-TCK26D634 was used. This antibody blocks thrombin- and plasmin-mediated TAFI activation, but not thrombin-thrombomodulin–mediated TAFI activation.19,20 TAFI zymogen levels were largely retained after injury in the presence of MA-TCK26D6, indicating that inhibition of thrombin- and/or plasmin-mediated TAFI activation prevented the consumption of TAFI zymogen after joint injury in FVIII−/− mice (Figure 4A-B). This excludes the thrombin-thrombomodulin complex as the main activator of TAFI after joint injury under these conditions. Furthermore, the virtually complete consumption of TAFI zymogen with recombinant human FVIII treatment points to a primary role for thrombin-mediated as opposed to plasmin-mediated TAFI activation after joint injury. Increased thrombin-antithrombin levels in aHA mice with anti-TAFI antibody compared with those in FVIII−/− mice also support added generation of thrombin in aHA to activate TAFI (supplemental Figure 7). It should be noted that TAFI is an acute-phase reactant in mice.39,40 Hence, TAFI levels may have risen above baseline with injury-induced bleeding, potentially masking partial activation by thrombin-thrombomodulin in the presence of MA-TCK26D6 sufficient for bleed protection. However, MA-TCK26D6 did increase joint bleeding in anti-FVIII antibody–treated WT mice (D2 Hct, 30% ± 5%; P < .05 vs baseline; supplemental Figure 8), similar to MA-RT36A3F5 (accelerating the inactivation rate of TAFIa; Figure 3A). MA-TCK26D6 is also capable of blunting the antifibrinolytic effect of TAFIa directly35 ; therefore, a potential contribution of TAFI activation by the thrombin-thrombomodulin complex cannot be formally excluded.
Vascular bed–specific contributions of TAFI deficiency to bleeding in HA
In agreement with previous studies, TAFI−/− mice did not demonstrate increased bleeding compared with WT mice within 20 minutes after tail resection (Figure 5A).16,41,42 Also, TAFI deficiency did not aggravate acute bleeding after tail resection in the presence of the anti-FVIII antibody (Figure 5A), suggesting that TAFI deficiency does not further increase rapid bleeding from the tail vasculature in aHA. To incorporate potential effects of TAFI on rebleeding events, Hct was measured 24 hours after tail resection as a surrogate measure for delayed bleeding. TAFI deficiency had no effect on Hct levels (Figure 5B). Because Hct at 24 hours only represented survivors, 24-hour mortality rates were determined. The 24-hour mortality rate was significantly increased from 0% to 50% in FVIII−/− mice and to 33% in anti-FVIII antibody–treated WT mice. TAFI deficiency caused no additional effects on mortality 24 hours after tail resection (Figure 5C; Table 1).
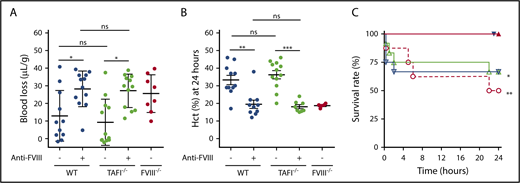
TAFI deficiency does not cause prolonged tail bleeding in HA mice. (A) Tail bleeding in WT C57Bl/6J, TAFI−/− C57Bl/6J, and FVIII−/− BALB/c mice with or without anti-FVIII antibody (0.25 mg/kg; n = 8-12). Bleeding was determined by measuring the mouse weight-normalized blood loss (µL/g) after tail resection for 20 minutes. (B) Bleeding determined by Hct at 24 hours after tail resection in WT C57Bl/6J, TAFI−/− C57Bl/6J, and FVIII−/− BALB/c mice, with or without anti-FVIII antibody (0.25 mg/kg), that survived 24 hours after tail resection. (C) Survival rate after tail resection of WT C57Bl/6J (n = 12; filled and open down triangles) or TAFI−/− C57Bl/6J (n = 12; filled and open up triangles) with (open triangles) or without (filled triangles) anti-FVIII antibody (0.25 mg/kg) and FVIII−/− BALB/c mice (n = 8; open circles). For the survival experiment, the log-rank (Mantel-Cox) test was used to compare experimental mice with WT mice after tail clip. Kruskal-Wallis test was used followed by Dunn’s post hoc multiple comparison test to compare experimental mice. *P < .05, **P < .01, ***P < .001. ns, not significant.
TAFI deficiency does not cause prolonged tail bleeding in HA mice. (A) Tail bleeding in WT C57Bl/6J, TAFI−/− C57Bl/6J, and FVIII−/− BALB/c mice with or without anti-FVIII antibody (0.25 mg/kg; n = 8-12). Bleeding was determined by measuring the mouse weight-normalized blood loss (µL/g) after tail resection for 20 minutes. (B) Bleeding determined by Hct at 24 hours after tail resection in WT C57Bl/6J, TAFI−/− C57Bl/6J, and FVIII−/− BALB/c mice, with or without anti-FVIII antibody (0.25 mg/kg), that survived 24 hours after tail resection. (C) Survival rate after tail resection of WT C57Bl/6J (n = 12; filled and open down triangles) or TAFI−/− C57Bl/6J (n = 12; filled and open up triangles) with (open triangles) or without (filled triangles) anti-FVIII antibody (0.25 mg/kg) and FVIII−/− BALB/c mice (n = 8; open circles). For the survival experiment, the log-rank (Mantel-Cox) test was used to compare experimental mice with WT mice after tail clip. Kruskal-Wallis test was used followed by Dunn’s post hoc multiple comparison test to compare experimental mice. *P < .05, **P < .01, ***P < .001. ns, not significant.
Mortality rate, proportion of deaths, and Hct at 24 h after tail resection
| Mortality rate, % (n/total) | P* | Hct, % | |
|---|---|---|---|
| WT | 0 (0/12) | 38 ± 7 | |
| TAFI−/− | 0 (0/12) | ns | 37 ± 7 |
| FVIII−/− | 50 (4/8) | <.01 | 19 ± 1 |
| WT + anti-FVIII | 33 (4/12) | <.05 | 20 ± 8 |
| TAFI−/− + anti-FVIII | 33 (4/12) | <.05 | 18 ± 3 |
| Mortality rate, % (n/total) | P* | Hct, % | |
|---|---|---|---|
| WT | 0 (0/12) | 38 ± 7 | |
| TAFI−/− | 0 (0/12) | ns | 37 ± 7 |
| FVIII−/− | 50 (4/8) | <.01 | 19 ± 1 |
| WT + anti-FVIII | 33 (4/12) | <.05 | 20 ± 8 |
| TAFI−/− + anti-FVIII | 33 (4/12) | <.05 | 18 ± 3 |
ns, not significant.
vs WT.
Therefore, in contrast to the observed increased bleeding after joint injury, TAFI deficiency did not increase bleeding after tail resection in mice treated with anti-FVIII antibody, indicating that the TAFI-mediated effects on bleeding were vascular bed specific and were of particular importance to the intra- and periarticular sites of the knee joints.
Differential effects of antifibrinolytic drugs on joint vs tail bleeding
Antifibrinolytic drug TXA was tested for its potential effects on joint bleeding to gain insight into the mechanism responsible for the ability of TAFIa to protect against joint bleeding in aHA. As a Lys analog, TXA impedes plasminogen binding to partially degraded fibrin. However, TXA did not correct injury-induced joint bleeding in either FVIII−/− mice (Figure 6A) or anti-FVIII antibody–treated TAFI−/− mice (Figure 6B). In contrast, TXA did correct tail bleeding in FVIII−/− mice (Figure 6C-D) and anti-FVIII antibody–treated WT mice (supplemental Figure 9), indicating that the TXA dosing used conveyed effective antifibrinolytic activity, thereby eliminating suboptimal dosing as a possible cause for the failure of TXA to inhibit joint bleeding.
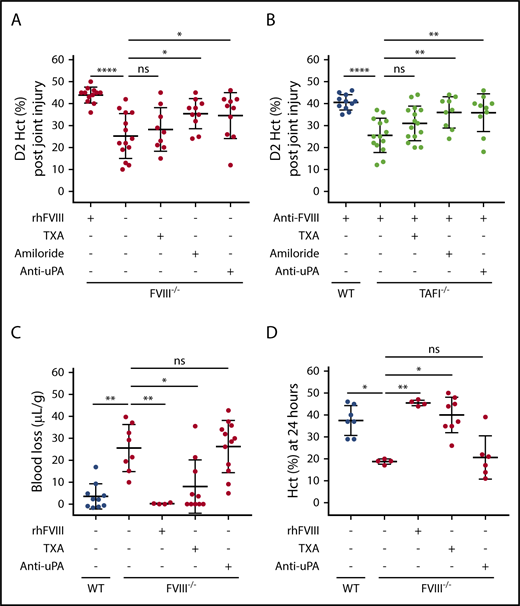
Differential effects of antifibrinolytic drugs on joint vs tail bleeding. Joint bleeding after knee injury in FVIII−/− BALB/c mice (n = 9-14) (A) or anti-FVIII antibody–treated (0.25 mg/kg) WT C57Bl/6J or TAFI−/− C57Bl/6J mice (n = 9-15) (B) upon treatment with recombinant human FVIII (rhFVIII; 200 IU/kg), TXA, amiloride, or anti-uPA antibody (10 mg/kg). Bleeding was determined by measuring Hct at day 2 (D2) postinjury. (C) Tail bleeding in FVIII−/− BALB/c mice (n = 4-12) upon treatment with rhFVIII (200 IU/kg), TXA, or anti-uPA antibody (10 mg/kg). Bleeding was determined by measuring the mouse weight-normalized blood loss (µL/g) after tail resection for 20 minutes. (D) Bleeding determined by Hct at 24 hours after tail resection in FVIII−/− BALB/c mice (n = 4-12) upon treatment with rhFVIII (200 IU/kg), TXA, or anti-uPA antibody (10 mg/kg). Kruskal-Wallis test was used followed by Dunn’s post hoc multiple comparison test to compare experimental mice. *P < .05, **P < .01, ****P < .0001. ns, not significant.
Differential effects of antifibrinolytic drugs on joint vs tail bleeding. Joint bleeding after knee injury in FVIII−/− BALB/c mice (n = 9-14) (A) or anti-FVIII antibody–treated (0.25 mg/kg) WT C57Bl/6J or TAFI−/− C57Bl/6J mice (n = 9-15) (B) upon treatment with recombinant human FVIII (rhFVIII; 200 IU/kg), TXA, amiloride, or anti-uPA antibody (10 mg/kg). Bleeding was determined by measuring Hct at day 2 (D2) postinjury. (C) Tail bleeding in FVIII−/− BALB/c mice (n = 4-12) upon treatment with rhFVIII (200 IU/kg), TXA, or anti-uPA antibody (10 mg/kg). Bleeding was determined by measuring the mouse weight-normalized blood loss (µL/g) after tail resection for 20 minutes. (D) Bleeding determined by Hct at 24 hours after tail resection in FVIII−/− BALB/c mice (n = 4-12) upon treatment with rhFVIII (200 IU/kg), TXA, or anti-uPA antibody (10 mg/kg). Kruskal-Wallis test was used followed by Dunn’s post hoc multiple comparison test to compare experimental mice. *P < .05, **P < .01, ****P < .0001. ns, not significant.
In addition to impeding plasminogen binding to fibrin and enhancing clot stability, TXA also considerably augments the ability of uPA to activate plasminogen, independent of fibrin, thereby increasing fibrinolysis.11,43,44 This dual role of TXA may explain its ineffectiveness in bleed reduction in inflamed tissues with high uPA expression such as in the synovial joint after bleeding.45,46 To probe a potential contribution of uPA-mediated activation of fibrinolysis to joint bleeding, uPA inhibitors were tested: amiloride, which is a clinically available antihypertensive molecule but also a potent inhibitor of uPA, and inhibitory anti-uPA antibody mU1.36 Amiloride reduced joint bleeding in FVIII−/− mice and anti-FVIII antibody–treated TAFI−/− mice significantly (Figure 6A-B). Antibody-mediated uPA blockade confirmed the contribution of uPA-induced plasmin formation to hemophilic joint bleeding (Figure 6A-B), because the antibody reduced joint bleeding in FVIII−/− mice (D2 Hct, 35% ± 10% vs 25% ± 10%; P < .05; Figure 6A) and anti-FVIII antibody–treated TAFI−/− mice (D2 Hct, 36% ± 9% vs 26% ± 8%; P < .01; Figure 6B). In contrast, neither amiloride nor the anti-uPA antibody reduced hemophilic tail bleeding (Figure 6C-D). Although these data suggest a uniform contribution of fibrinolysis to bleeding, either after joint injury or tail resection, the difference in the plasminogen activators responsible for initiation of fibrinolysis (uPA in the joint but not in the tail and tPA in the tail) emphasizes differential vascular bed–specific contributions of plasminogen activators.
Discussion
The different bleeding diathesis of congenital and acquired hemophilias is clinically well recognized but mechanistically poorly understood.6,47,48 Reproducing a notably milder joint injury–induced bleeding phenotype in aHA WT mice compared with FVIII−/− mice provided a clinically relevant model to unravel hemostatic mechanisms specific to the joints. As opposed to the mild joint bleeding in aHA mice, tail bleeding was severe, indicating that aHA compromises coagulation in vivo severely. This finding is in agreement with the ex vivo effect of the anti-FVIII antibody in mouse plasma shown here and in normal human plasma in vitro.18 The joint-specific protection against bleeding in the presence of the anti-FVIII antibody may therefore be tied to antifibrinolytic mechanisms and clot protection rather than clot formation and influenced by substrates sensitive to variations in thrombin generation, such as TAFI. Indeed, the anti-FVIII antibody dose dependently reduced thrombin generation in mouse plasma ex vivo, whereas TAFI activation and clot lysis acceleration were considerably less affected by the anti-FVIII antibody.
TAFI gene deficiency increased joint bleeding substantially in aHA, such that it was indistinguishable from that in congenital HA mice. This implies that TAFI protects from severe joint bleeding in aHA and suggests that joint bleeding in congenital hemophilia might be exacerbated by defective TAFI activation. Indeed, more than a decade of in vitro studies have demonstrated that TAFI activation is defective in hemophilia plasma, but in vivo evidence is lacking.14,15,21,49 Here, we demonstrate that TAFI activation in vivo was significantly impaired after joint injury in congenital FVIII deficiency but not in aHA. Therefore, TAFIa is a modifier of phenotypic joint bleeding in hemophilia, with the degree of TAFI activation inversely correlating with the severity of bleeding after joint injury.
The defective activation of TAFI in FVIII−/− mice after joint injury provides important novel insights into the physiological activation mechanism of TAFI in vivo. Thrombomodulin enhances TAFI activation by thrombin >1000-fold and overcomes the need for FXIa-mediated intrinsic pathway amplification to generate sufficient thrombin for TAFI activation. Therefore, thrombin-thrombomodulin has often been portrayed as the physiological activation mechanism for TAFI in vivo.31 In the hemophilic joint, however, inhibition of TAFI activation by the activation pathway–selective antibody (MA-TCK26D6)34,35 suggests that thrombin-mediated TAFI activation was the relevant hemostatic mechanism after joint injury, independent of thrombomodulin despite its reported relative abundance on synoviocytes.23,50
Consistent with the upregulation of uPA levels by monocyte and macrophage recruitment after joint injury in FVIII−/− mice, inhibition of uPA corrected the loss of TAFIa-dependent joint bleeding protection in aHA TAFI−/− mice. In addition, TAFIa mediates a host of antiinflammatory activities by modulating proinflammatory mediators, such as C3a, C5a, bradykinin, and others, and is specifically beneficial in dampening chronic inflammation associated with joint diseases, such as rheumatoid and osteoarthritis.51,52 Furthermore, the TAFI activation defect in hemophilia and resulting inability of TAFIa to moderate the chemoattractant activity of C5a may facilitate the recruitment of uPA-secreting inflammatory cells to the injured joints, further aggravating bleeding. Thus, the difference in joint bleeding susceptibility between acquired and congenital hemophilia provides an emerging concept that assigns a critical role to uPA-induced bleeding controlled by TAFIa. In congenital FVIII deficiency, the fibrinolytic response continues unchecked because of severely defective TAFI activation, resulting in aggravated bleeding. Partial activation of TAFI in aHA provides a sufficiently strong brake on uPA-induced activation of fibrinolysis, thereby lowering the susceptibility of joint bleeding in aHA.
In comparing the tail transection and joint injury bleeding models, clear differences were found in the mechanisms responsible for severe bleeding. The diverging bleeding patterns in the presence of a uPA inhibitor or TXA revealed that the plasminogen activator involved in bleeding was vascular bed specific, delineating bleeding in the joint to be primarily driven by uPA and tail bleeding by tPA. Although TXA may also have other effects, the involvement of tPA in tail bleeding is based on the interpretation that TXA primarily inhibits tPA-mediated fibrinolysis. Therefore, differences in joint vs tail bleeding originate in part from vascular bed–specific fibrinolysis.
However, the different plasminogen activating mechanisms do not explain why aHA WT mice were protected against joint bleeding but not against tail bleeding. Vascular bed–specific contributions of endogenous TAFI also take part in the mechanistic divide between joint and tail bleeding. This is evidenced by the fact that the endogenous TAFI system protected the joint against bleeding after injury in WT aHA mice but did not convey bleed protection after tail transection, in agreement with previous reports.41,42 However, administration of exogenous recombinant TAFI could significantly reduce tail bleeding in FVIII−/− mice, suggesting that tPA-mediated fibrinolysis in the tail vasculature contributes to bleeding.53 Additionally, the lack of a hemostatic effect in the tail vasculature by endogenous TAFI indicates that TAFIa is not sufficiently generated to overcome the threshold level to exert its antifibrinolytic effect and protect against tail bleeding (Figure 7).
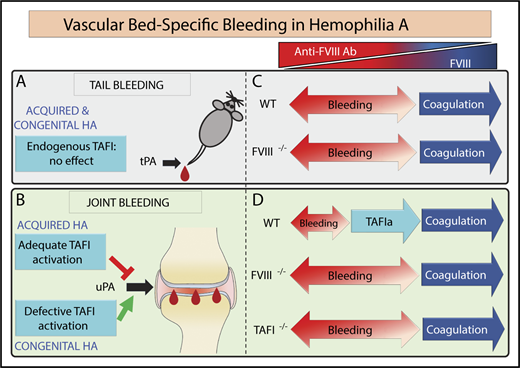
Proposed mechanism of vascular bed–specific bleeding in aHA vs congenital HA. The congenital HA mouse is characterized by excessive tail and joint bleeding. In contrast, the aHA mouse featured a divergent bleeding phenotype existing of excessive tail bleeding but mild joint bleeding. Therefore, the divergent bleeding was vascular bed specific, encompassing different fibrinolytic mechanisms. (A) Endogenous TAFI did not control tail bleeding in either aHA or congenital HA mice, because tail bleeding in aHA was not exacerbated by TAFI gene deficiency. In contrast, antifibrinolytic treatment with exogenous TAFI53 or TXA was able to restore hemostasis after tail resection, suggesting that tPA-driven hyperfibrinolysis contributed to hemophilic tail bleeding. (B) In the joints, the extent of bleeding diverged between aHA (minimal bleeding) vs congenital HA (excessive bleeding), and this difference in bleeding severity was due to the extent of TAFI activation. Residual thrombin formation in aHA provided adequate TAFI activation for bleed protection, but the defective TAFI activation in congenital HA was not associated with bleed protection and instead contributed to severe bleeding. Hemophilic joint bleeding was primarily mediated by uPA and largely unresponsive to treatment with TXA, in contrast to hemophilic tail bleeding. (C-D) Primary mechanisms responsible for achieving hemostasis (coagulation vs antifibrinolysis by TAFIa) for each type of bleeding (tail vs joint) depending on the level of FVIII (or the inverse anti-FVIII antibody level). (C) No differences were observed in the extent of tail bleeding between congenital HA (FVIII−/−) and aHA WT mice (even when treated at a low dose of anti-FVIII antibody). (D) In contrast, joint bleeding in aHA WT mice was minimal but became severe upon elimination of TAFI. Moreover, aHA TAFI−/− mice and congenital HA mice bled to the same extent after joint injury. Therefore, the hemostasis gap between aHA and congenital HA in the joint was due to adequate vs defective activation of TAFI, respectively. This indicated that residual thrombin generation in aHA was insufficient to induce functional coagulation to arrest bleeding but proficient to activate TAFI and achieve hemostasis in the joints. This study points to an important modifying role for TAFI in joint bleeding in hemophilia.
Proposed mechanism of vascular bed–specific bleeding in aHA vs congenital HA. The congenital HA mouse is characterized by excessive tail and joint bleeding. In contrast, the aHA mouse featured a divergent bleeding phenotype existing of excessive tail bleeding but mild joint bleeding. Therefore, the divergent bleeding was vascular bed specific, encompassing different fibrinolytic mechanisms. (A) Endogenous TAFI did not control tail bleeding in either aHA or congenital HA mice, because tail bleeding in aHA was not exacerbated by TAFI gene deficiency. In contrast, antifibrinolytic treatment with exogenous TAFI53 or TXA was able to restore hemostasis after tail resection, suggesting that tPA-driven hyperfibrinolysis contributed to hemophilic tail bleeding. (B) In the joints, the extent of bleeding diverged between aHA (minimal bleeding) vs congenital HA (excessive bleeding), and this difference in bleeding severity was due to the extent of TAFI activation. Residual thrombin formation in aHA provided adequate TAFI activation for bleed protection, but the defective TAFI activation in congenital HA was not associated with bleed protection and instead contributed to severe bleeding. Hemophilic joint bleeding was primarily mediated by uPA and largely unresponsive to treatment with TXA, in contrast to hemophilic tail bleeding. (C-D) Primary mechanisms responsible for achieving hemostasis (coagulation vs antifibrinolysis by TAFIa) for each type of bleeding (tail vs joint) depending on the level of FVIII (or the inverse anti-FVIII antibody level). (C) No differences were observed in the extent of tail bleeding between congenital HA (FVIII−/−) and aHA WT mice (even when treated at a low dose of anti-FVIII antibody). (D) In contrast, joint bleeding in aHA WT mice was minimal but became severe upon elimination of TAFI. Moreover, aHA TAFI−/− mice and congenital HA mice bled to the same extent after joint injury. Therefore, the hemostasis gap between aHA and congenital HA in the joint was due to adequate vs defective activation of TAFI, respectively. This indicated that residual thrombin generation in aHA was insufficient to induce functional coagulation to arrest bleeding but proficient to activate TAFI and achieve hemostasis in the joints. This study points to an important modifying role for TAFI in joint bleeding in hemophilia.
The protection of TAFI against bleeding and the profibrinolytic mechanisms that contribute to bleeding in hemophilia were found to be vascular bed-specific, thereby giving credence to the term vascular bed–specific bleeding in extension of the paradigm vascular bed–specific thrombosis. Appreciation of mechanistic differences in vascular bed–specific bleeding is key for designing improved antibleeding strategies for hemophilia but may also help with the interpretation of the effectiveness of current therapies. For instance, TXA is effectively used to reduce surgery-induced bleeding in nonhemophilia patients after joint replacement, presumably because of large-vessel bleeding associated with surgery. In contrast, its efficacy in hemophilic joint bleeding, presumably caused by synovial microvessel bleeding, remains elusive.54,55 However, TXA is effective as adjuvant treatment to clotting factor (or bypassing agent) against mucosal bleeding or dental extractions.54,55 In this regard, it is important to note that in contrast to inhibition of tPA-mediated fibrinolysis, TXA promotes uPA-mediated plasminogen activation, and TXA has been reported to worsen delayed intracranial hemorrhaging after mouse traumatic brain injury, which has been attributed to the shift from tPA- to uPA-mediated fibrinolysis.11,43,44,56
In summary, this study unmasked a prominent role for endogenous TAFI in the protection of the vascular bed of the joint against bleeding that is at least in part responsible for the notably milder phenotypic joint bleeding in aHA compared with congenital HA. This protective mechanism is dysfunctional in congenital HA mice because of the inherent defect in thrombin generation that is required for the activation of TAFI in the joint. Therefore, clot protection strategies that restore TAFI activation are important to blunt the susceptibility of the hemophilic joint to bleeding.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by a National Hemophilia Foundation Judith Graham Pool Research Fellowship (T.W., E.J.C.), an American Heart Association Western States Postdoctoral Fellowship (T.W.), a National Hemophilia Foundation Career Development Award (A.v.D.), and National Institutes of Health, National Heart, Lung, and Blood Institute grants HL104165, HL130678, and HL142975 (L.O.M.).
Authorship
Contribution: T.W. designed and performed the research, analyzed and interpreted the data, performed statistical analysis, and wrote the manuscript; E.J.C. contributed to tail clip experiments; P.J.D., N.B., and J.C.M.M. provided unique materials and mice; A.v.D. provided scientific and experimental guidance; L.O.M. conceived and designed the study, provided project oversight and experimental guidance, and wrote the manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: A.v.D. has received honoraria for participating in scientific advisory board panels, consulting, and speaking engagements for Baxalta/Shire, Bayer, Pfizer, Bioverativ (now Sanofi), CSL-Behring, and Novo Nordisk. The employer of J.C.M.M. received consultancy fees from Bayer and Daiichi Sankyo. The remaining authors declare no competing financial interests.
Correspondence: Laurent O. Mosnier, Department of Molecular Medicine (IMM-315), The Scripps Research Institute, 10550 North Torrey Pines Rd, La Jolla, CA 92037; e-mail: lmosnier@scripps.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal