

Sickle cell trait (SCT) is unique among the carrier states that are identified during newborn screening. Unlike other heterozygous states for rare recessive diseases, SCT is exceedingly prevalent throughout regions of the world, making sickle cell disease one of the most common monogenetic diseases worldwide. Because of this high frequency, reproductive counseling is of paramount importance. In addition, unlike other carrier states, SCT seems to be a risk factor for several clinical complications, such as extreme exertional injury, chronic kidney disease, and venous thromboembolism. Increasing knowledge about these clinical outcomes can help inform genetic counseling recommendations. Expanding research and clinical efforts are needed to ensure that the promises of modern and precision medicine can be delivered to the millions of SCT carriers and their children.
Introduction
Sickle cell trait (SCT) is one of the most common hemoglobin mutations in the world because of its protective effects against severe malaria. In the United States, SCT is found in nearly 3 million individuals, constituting 7% to 9% of the African American population, and worldwide, the number of individuals with SCT exceeds 300 million, with prevalence rates that can exceed 25% in regions where malaria is endemic, such as Nigeria and tribal India.1,2 The evolutionary advantage of SCT is profound and undisputed, conferring a remarkable 90% risk reduction against severe and cerebral malarias.3
The high prevalence of SCT also leads to several reproductive counseling and clinical management issues that are relevant to patients, primary care physicians, and general hematologists. SCT carriers are at risk for having children with sickle cell disease (SCD); therefore, pre- and postconception counseling is of significant importance. In addition, although SCT has largely been considered a benign condition, it has been found to be a risk factor for several clinical outcomes, ranging from the rare complications of exertion-related injury and renal medullary carcinoma (RMC) to more common medical conditions, such as chronic kidney disease (CKD) and venous thromboembolism (VTE); therefore, knowledge about the current epidemiologic evidence can help guide genetic counseling recommendations for SCT carriers. This review will highlight both the reproductive and clinical implications of SCT and provide a framework for counseling recommendations for individuals with SCT.
Notification and reproductive counseling practices for SCT carriers
Screening for SCT
Newborn screening (NBS) programs have been developed in several countries to allow for the early identification of infants with SCD and facilitate the institution of early life-saving treatments such penicillin prophylaxis.1 In the United States, mandatory universal NBS has been available in the majority of states since 1990 and in all 50 states since 2006.4 However, in much of Europe, optional or targeted NBS programs for at-risk racial populations are the predominant standard. In England, for example, the National Health Service established an NBS program in 2006, which offers optional screening at 5 to 8 days of life for all infants.5 Although most parents do provide consent for screening, no protocols currently exist for testing children whose parents decline testing.6 Similarly, other European countries, such as France and Belgium, have adopted targeted national NBS programs. These programs are designed to screen newborns who are either deemed to be from high-risk racial groups7 or born in select regions with a high density of at-risk groups.8 In India and Africa, where SCT carrier rates are highest, national NBS programs do not yet exist, but many promising regional and national pilot programs are emerging.9,,-12
Although SCT is a recognized global public health concern because of its significant reproductive and clinical implications,13 there are substantial challenges related to screening for SCT carriers through NBS. As described, in countries with the highest prevalence of SCT, the potential of universal NBS has not yet been realized. Furthermore, in countries where targeted NBS is performed, correct identification of at-risk parents and infants relies on patient self-knowledge and/or health care providers’ knowledge of the diverse geographic origins of SCT, both of which are often inaccurate.14,15 In the United States, because identification of SCT is a byproduct of NBS for SCD, standards for notification and documentation of SCT status have not been established. Notification of SCT carrier results through NBS differs widely by state, with the majority of states providing results to pediatricians but not directly to parents.16 Because guidelines for counseling by pediatricians do not exist, how information about SCT is communicated to families, whether this information is retained in the medical record over a lifetime, and whether parents pass information on SCT to their children are not known.
In higher-income countries, electronic medical records have been suggested as a method to help retain SCT status as part of a child’s medical record from infancy into adulthood; however, obstacles to maximizing the potential of this technology include the many sickle cell–related diagnostic codes that lead to errors in identifying patients with SCD vs SCT. In the United States, medical records do not transfer across health care systems, and information about patients’ SCT status may be lost when patients transition between providers. Legislative and funding initiatives to allow for expansion of existing NBS programs have also been considered to allow states to more effectively notify individuals and parents about SCT screening results. For example, a few states, such as California, have introduced legislation to support NBS programs to develop standard protocols for renotification and dissemination of SCT status information to individuals as they reach reproductive age; however, funding for such programs has thus far been limited.17
Because NBS programs were not initially designed for identification and notification of carrier states, it is not surprising that the aforementioned limitations exist. As knowledge about SCT status becomes a priority, efforts should be made to extend existing programs to provide universal NBS in countries where only targeted screening is performed and to improve notification practices and medical record documentation for individuals who screen positive for SCT.
Mass reproductive counseling efforts in SCT
Premarital counseling of SCT carriers has been considered as a strategy to reduce SCD prevalence, because such efforts have proven successful in reducing rates of severe β-thalassemia phenotypes in small regions of high endemicity such as Cyprus and Sardinia.18 Worldwide, efforts at primary prevention have ranged from legally mandated premarital hemoglobinopathy testing to public education campaigns. Although premarital counseling and education have historically been considered effective strategies for reducing autosomal recessive disease, especially in regions where arranged marriage is customary, changing cultural norms and preferences have hampered the success of such programs in the modern era. For example, in Saudi Arabia, a 2003 royal decree mandated premarital screening for β-thalassemia and SCD before the drafting of a marriage contract; however, subsequent studies found that premarital testing did not meaningfully change marriage or reproductive decision making. Specifically, among 934 couples identified as being at risk for having a child with β-thalassemia or SCD in Saudi Arabia, 88% of couples with SCT still chose to marry.14 Similar results have been demonstrated in studies in the United States and Jamaica. In the United States, a randomized controlled study of an electronic educational platform for young adults with SCT demonstrated a significant increase in participant knowledge of SCT and SCD but did not affect participant reproductive decision making.15 In Jamaica, a large study of a comprehensive SCT educational program conducted among 16 600 high school seniors failed to reduce the rate of SCD births in the participants after 8 years of follow-up.19
Although these studies suggest that mass premarriage or preconception educational campaigns may not be efficacious in the modern era, long-term studies to determine the impact of such programs on SCD birth rates in multiple different populations have not been performed. Furthermore, the success of public or targeted educational programs on the behavior of more mature adults, rather than adolescents, who may have delayed childbearing into their later reproductive years has not been explored.
Targeted pre- and postconception counseling in SCT
Identification of hemoglobinopathy carrier status during the pregnancy planning or postconception stage is important to help guide counseling of women and their partners. In the United States, the American College of Obstetrics and Gynecology recommends targeted pre- and postconception screening of those of African, Southeast Asian, and Mediterranean decent for hemoglobinopathy carrier status.20 Despite these recommendations, surveys of American College of Obstetrics and Gynecology fellows have demonstrated that screening practices vary widely and that knowledge of hemoglobinopathy assessment among obstetricians is generally low.21 Nonetheless, screening allows providers to present at-risk patients with several diagnostic and reproductive technologies to guide their pregnancy decisions. Figure 1 provides a decision tree that highlights the opportunities for the use of preimplantation genetic diagnosis and fetal diagnostics, based on patient preference and values.
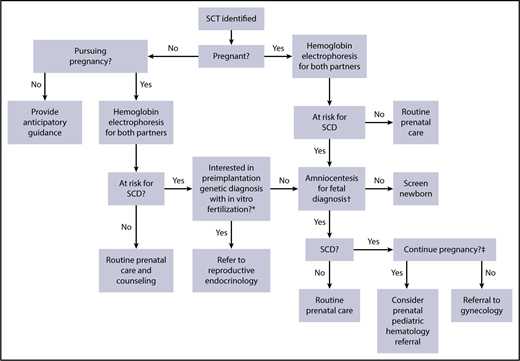
Reproductive decision-making tree for SCT carriers and their physicians. Each branch in this decision tree is contingent on test or procedural availability and accessibility, and decisions are contingent on patient preferences and values. *Preimplantation genetic diagnosis may not be available because of patient location and prohibitive cost. †Chorionic villus sampling or amniocentesis for fetal diagnosis is performed as early as 11 weeks of gestation and offered through week 20; results from this testing may take 1 to 2 weeks to return. ‡Decisions to terminate pregnancy may be affected by insurance and legal restrictions.
Reproductive decision-making tree for SCT carriers and their physicians. Each branch in this decision tree is contingent on test or procedural availability and accessibility, and decisions are contingent on patient preferences and values. *Preimplantation genetic diagnosis may not be available because of patient location and prohibitive cost. †Chorionic villus sampling or amniocentesis for fetal diagnosis is performed as early as 11 weeks of gestation and offered through week 20; results from this testing may take 1 to 2 weeks to return. ‡Decisions to terminate pregnancy may be affected by insurance and legal restrictions.
Preimplantation genetic diagnosis (PGD) used in conjunction with in vitro fertilization (IVF) for SCD was first reported in 1999.22 For at-risk parents, PGD facilitates selective introduction of embryos unaffected by SCD into the uterus. Recent advances in PGD technology, such as small-volume biopsy techniques, have improved embryo safety, diagnostic accuracy, and cost effectiveness of PGD. Furthermore, single-cell genomic and noninvasive sampling techniques are currently in development, which promise to further improve diagnostics and reduce potential ethical concerns.23
Although the existence of this technology may expand the reproductive options for SCT carriers, there are 3 important preconditions that limit the use of PGD in many settings: awareness of the hemoglobinopathy status of both partners, planned pregnancy, and ability to pay for the procedure. As described in the previous section, many individuals are not aware of their own or their partners’ SCT status and are also either unaware or uninformed about their carrier status for other at-risk β-globin variants, such as hemoglobin C trait or β-thalassemia. In addition, the use of PGD requires planned pregnancy, the rates of which differ by race, education, and cultural practices. In the United States, 45% of all pregnancies are unintended, with unplanned pregnancy rates >50% in black women, who are disproportionately at risk for having children with SCD.24 Moreover, PGD is mostly available in high-income settings because of the required technological expertise and the high health care costs associated with IVF. In the United States, the median out-of-pocket cost for 1 cycle of IVF is $24 000 and for a successful pregnancy is $61 000.25 Because there are no standards for insurance coverage of PGD among SCT carriers in the United States, these costs may be prohibitive.
Worldwide, universal health care systems in high-income countries may allow the introduction of national coverage of PGD for couples at risk of having a child with SCD. England’s National Health Service, for example, has permitted 3 cycles of PGD to at-risk women with hemoglobinopathies since 2004. However, despite its availability, PGD is not often offered or ultimately performed.26 In 1 study conducted between 2002 and 2007, only 78 couples from southeastern England were referred for consideration of PGD to prevent SCD. Ultimately, 12 individuals pursued the intervention, resulting in a total of 16 IVF cycles, 4 pregnancies, and 2 live births.27 Notably, the mean number of oocytes collected, oocyte fertilization rate, and live birth rate were lower among couples pursuing potential diagnosis of SCD compared with other autosomal recessive disorders in this study; however, the reasons for this have not been fully elucidated. In the United States, data on outcomes and acceptability of PGD among SCT parents are limited, although a few small studies have demonstrated that most individuals with SCT reported that they had not been previously educated about PGD and believed that information about PGD should be routinely offered as part of reproductive counseling for at-risk parents.28 These data suggest that education about PGD is largely overlooked by providers and that PGD is likely underutilized among SCT carriers.
Unlike PGD, which relies on pregnancy planning, chorionic villus sampling (gestational age, 10-13 weeks) and amniocentesis (gestational age, 15-20 weeks) provides options for antenatal diagnosis of SCD after conception. Although noninvasive prenatal diagnostic techniques, such as fetal cell-free DNA isolated from maternal blood, are in development, they are not yet available for the diagnosis of SCD.29 The decision about whether to perform invasive prenatal diagnostics requires an in-depth discussion with the pregnant woman and/or partner regarding their views about continuation of an affected pregnancy. If prenatal diagnosis is pursued, avoiding delays in obtaining and communicating results is imperative, because the medical options and availability of pregnancy termination are reduced if diagnosis is delayed well into the second trimester. Early referral to a pediatric hematologist should be considered when a prenatal diagnosis of SCD is established.
Importantly, in the case of both PGD and prenatal screening, counseling must be individualized and patients’ values and their preferences carefully explored. Research suggests that SCT carriers may not view SCD as life threatening or may perceive SCD as a manageable disorder; therefore, risk-based counseling must take into account parents’ experiences and perceptions of SCT and SCD.30 However, providers cannot assume that preferences are homogenous in the diverse population of those who carry SCT. For example, 1 study found that women in Cameroon who learned their fetus was affected by SCD were more willing than their providers expected to terminate their pregnancy.31 Ultimately, these life-changing reproductive decisions are deeply personal and may be influenced by individual, cultural, or religious beliefs regarding the significance of having a child with SCD and the ethics of artificial reproductive technologies and abortion.
Clinical complications of SCT
SCT is unique among carrier states identified during NBS, not only because of its high prevalence and implications for reproductive counseling, but also because it has been identified as a risk factor for several clinical complications. The recent surge in epidemiologic research has provided much-needed data about the risk and modifying factors for SCT-related clinical outcomes and can help inform genetic counseling recommendations for SCT carriers (Table 1).
Major clinical complications of SCT and proposed counseling recommendations
| Complication | Recommendations for counseling |
|---|---|
| Exertion-related injury | The vast majority of individuals with SCT will never experience an exertion-related injury event |
| Even among those involved in high-intensity competitive sports or training, the absolute risk of exertional injury with SCT is low | |
| Exertional injury in SCT does not seem to occur with usual exercise or low- to moderate-intensity exertion | |
| Modifiable factors such as activity duration/recovery, hydration, and climate acclimation likely influence the risk of exertional injury in SCT | |
| Universal precautions, such as those employed by the military,* seem to mitigate the risk of exertional injury in all individuals, irrespective of SCT status | |
| Screening for SCT, even in high-intensity settings, is not necessary, especially when universal precautions are instituted | |
| CKD | CKD occurs in ∼15%-35% of adults with SCT age >45 y, which is approximately twice the risk of those without SCT |
| The risk of progression to ESRD in carriers of SCT is unknown | |
| Not all SCT carriers are at risk for CKD, and those with coinheritance of α-thalassemia mutations may be protected from developing CKD | |
| Modifiable factors such as coexisting diabetes may influence the risk of CKD in SCT | |
| Therapies for the prevention and treatment of CKD in SCT are unknown | |
| Research is needed to determine the benefit of screening SCT carriers for CKD | |
| RMC | RMC is an exceedingly rare cancer; therefore, the vast majority of individuals with SCT will never develop RMC |
| RMC can occur in children or adults, with ∼50% of cases occurring at <21 y of age | |
| Hemoglobinopathy testing should be performed in any child or young adult with micro- or macroscopic hematuria | |
| Prompt referral to urology or renal imaging should be performed in any individual with SCT and unexplained hematuria | |
| Routine screening for RMC among known SCT carriers without concerning symptoms is not recommended given the rarity of the diagnosis | |
| VTE | SCT is a low-risk thrombophilia, similar to factor V Leiden or prothrombin gene mutation |
| Screening for SCT in individuals with VTE is not recommended, and its presence should not influence anticoagulation duration | |
| D-dimer results in SCT carriers should be interpreted with caution and may not be reliable for guiding anticoagulation discontinuation or ruling out VTE | |
| Stroke | SCT does not seem to be a risk factor for ischemic stroke |
| SCT should not be assumed to be the etiology of stroke in an SCT carrier, and additional workup for underlying cause should be pursued |
| Complication | Recommendations for counseling |
|---|---|
| Exertion-related injury | The vast majority of individuals with SCT will never experience an exertion-related injury event |
| Even among those involved in high-intensity competitive sports or training, the absolute risk of exertional injury with SCT is low | |
| Exertional injury in SCT does not seem to occur with usual exercise or low- to moderate-intensity exertion | |
| Modifiable factors such as activity duration/recovery, hydration, and climate acclimation likely influence the risk of exertional injury in SCT | |
| Universal precautions, such as those employed by the military,* seem to mitigate the risk of exertional injury in all individuals, irrespective of SCT status | |
| Screening for SCT, even in high-intensity settings, is not necessary, especially when universal precautions are instituted | |
| CKD | CKD occurs in ∼15%-35% of adults with SCT age >45 y, which is approximately twice the risk of those without SCT |
| The risk of progression to ESRD in carriers of SCT is unknown | |
| Not all SCT carriers are at risk for CKD, and those with coinheritance of α-thalassemia mutations may be protected from developing CKD | |
| Modifiable factors such as coexisting diabetes may influence the risk of CKD in SCT | |
| Therapies for the prevention and treatment of CKD in SCT are unknown | |
| Research is needed to determine the benefit of screening SCT carriers for CKD | |
| RMC | RMC is an exceedingly rare cancer; therefore, the vast majority of individuals with SCT will never develop RMC |
| RMC can occur in children or adults, with ∼50% of cases occurring at <21 y of age | |
| Hemoglobinopathy testing should be performed in any child or young adult with micro- or macroscopic hematuria | |
| Prompt referral to urology or renal imaging should be performed in any individual with SCT and unexplained hematuria | |
| Routine screening for RMC among known SCT carriers without concerning symptoms is not recommended given the rarity of the diagnosis | |
| VTE | SCT is a low-risk thrombophilia, similar to factor V Leiden or prothrombin gene mutation |
| Screening for SCT in individuals with VTE is not recommended, and its presence should not influence anticoagulation duration | |
| D-dimer results in SCT carriers should be interpreted with caution and may not be reliable for guiding anticoagulation discontinuation or ruling out VTE | |
| Stroke | SCT does not seem to be a risk factor for ischemic stroke |
| SCT should not be assumed to be the etiology of stroke in an SCT carrier, and additional workup for underlying cause should be pursued |
ESRD, end-stage renal disease.
More information provided in Table 2.
Exertion-related injury
Perhaps the most well known and debated complication of SCT is exertional injury. Reports of sudden death and rhabdomyolysis both in the military and among competitive athletes in the National Collegiate Athletic Association (NCAA) have led to significant concerns about the potential severe risk associated with extreme exertion in SCT carriers. Although evidence for these severe complications is largely anecdotal, several large studies have provided important insights that can be incorporated into genetic counseling guidelines.
Sudden death is both the most rare and feared potential complication of SCT. The first large population-based study of sudden death in those with SCT was performed among military personnel from 1977 to 1981, demonstrating a nearly 15-fold increased relative risk of sudden death among the African American recruits.32 Although a seemingly staggering risk, the estimate was based on only a small number of deaths, 13 (0.03%) among SCT carriers and 10 (0.002%) among non-SCT carriers. Studies among athletes in the NCAA have also demonstrated a large relative risk (approximately fivefold) but low absolute risk (0.02%) of exertional death among African American SCT carriers.33 These reports, therefore, suggest that only a small fraction of individuals with SCT are ultimately at risk for this severe complication and that modifying factors are important in determining and mitigating risk.
Likely the most important of these modifying factors is intensity of exertion. It is important to note that the studies of sudden death were performed in select groups of young adults who participated in extreme physical exertion. These findings, therefore, cannot be extrapolated to the general population, where sudden death is often a result of an underlying chronic condition, such as cardiac disease or arrhythmia. In addition, usual exertion patterns, such as with recreational exercise or sport, are not likely to result in sudden death among SCT carriers. In fact, even among NCAA athletes, most sudden death events in SCT were found to have occurred during intense practice sessions (not games themselves) for Division I football alone, a finding that attests to the environmental extremes underlying this risk.33
Additional factors such as activity duration/recovery, hydration, and climate acclimation likely also significantly contribute to exertional injury with high-intensity exercise. In 2003, the US Army formalized a policy of universal precautions to mitigate risk of exertion-related injury among all of its recruits.34 This policy, which includes various leader, clinician, and individual prevention measures for exertional collapse, includes such risk controls as limiting high-intensity training to cool mornings and avoiding high-intensity activity on consecutive days (Table 2). The institution of these measures has been largely successful, resulting in a decrease in sudden death both in SCT and non-SCT carriers. Notably, a recent landmark study of ∼50 000 African American army recruits from 2011 to 2014 did not find an increased risk of nonbattle death among those with SCT.34 Rates of mild heat injury and heat stroke were similarly not higher among SCT carriers in this modern army cohort.35 These epidemiologic data support the use of universal precautions to prevent exertional injury with all high-intensity training, irrespective of SCT status, and suggest that SCT screening is unnecessary in these contexts.
Adapted summary of the US Army universal precautions protocol34
| Leader and individual prevention measures |
|---|
| Identify and assess hazards |
| Check recent and anticipated environmental conditions and associated heat stress levels |
| Determine unique group and individual risk factors |
| Use the HEAT acronym: |
| Heat levels and associated risk |
| Exertion levels planned |
| Acclimation level of those present for training |
| Time factors including duration of activity and recovery time |
| Develop and implement risk controls |
| Plan training in advance and estimate risk of collapse events, taking into account: |
| Training event characteristics |
| Uniform and equipment needed |
| Location and time of day of key activities |
| Institute the following protocols according to hazard identified: |
| Adjust activity distances, duration, pace, and loads |
| Include work/rest cycles |
| Conduct high-intensity training in cooler morning hours |
| Ensure no consecutive days of high-intensity activity |
| Ensure proper resources available: |
| Equipment for checking wet-bulb globe temperatures (measure of heat stress in direct sunlight, taking into account temperature, humidity, wind speed, sun angle, and cloud cover) |
| Water, snacks, and electrolyte beverages |
| Medical resources and communication capabilities |
| Establish standard operating procedures, tested on soldiers |
| Leader and individual prevention measures |
|---|
| Identify and assess hazards |
| Check recent and anticipated environmental conditions and associated heat stress levels |
| Determine unique group and individual risk factors |
| Use the HEAT acronym: |
| Heat levels and associated risk |
| Exertion levels planned |
| Acclimation level of those present for training |
| Time factors including duration of activity and recovery time |
| Develop and implement risk controls |
| Plan training in advance and estimate risk of collapse events, taking into account: |
| Training event characteristics |
| Uniform and equipment needed |
| Location and time of day of key activities |
| Institute the following protocols according to hazard identified: |
| Adjust activity distances, duration, pace, and loads |
| Include work/rest cycles |
| Conduct high-intensity training in cooler morning hours |
| Ensure no consecutive days of high-intensity activity |
| Ensure proper resources available: |
| Equipment for checking wet-bulb globe temperatures (measure of heat stress in direct sunlight, taking into account temperature, humidity, wind speed, sun angle, and cloud cover) |
| Water, snacks, and electrolyte beverages |
| Medical resources and communication capabilities |
| Establish standard operating procedures, tested on soldiers |
Adapted from Nelson et al.34
Limited data, however, do demonstrate a persistent moderate (1.5-fold) independent risk of exertional rhabdomyolysis among SCT carriers in the military that is not completely mitigated by universal precautions.34 Similar to sudden death, rhabdomyolysis is a rare complication; therefore, only a small proportion of SCT carriers (1.2%) are ultimately at risk, and additional modifying factors, such as coinheritance of genetic variant(s), may be contributory. Importantly, factors such as obesity, tobacco use, and medication use demonstrate a higher independent risk for rhabdomyolysis than SCT and are easily modifiable targets for risk mitigation in those engaged in high-intensity exertion.
Splenic infarction also seems to be a rare complication observed in SCT carriers based on numerous case reports and series. The major risk factor for splenic infarction in SCT has been noted to be altitude, with a majority of cases being reported at 5000 to 7500 feet above sea level.36 Athletes performing high-intensity exercise at high altitudes have also been reported to develop splenic infarction, suggesting that exertion and dehydration may exacerbate the risk.37
Unlike in the military, where universal precautions have long been well established, protocols to reduce exertional injury in the NCAA have been slow to be adopted.38 A National Athletic Trainers’ Association task force issued a position paper in 2012 that outlined proposed guidelines to decrease sudden death rates during collegiate conditioning sessions, which included activity acclimation, appropriate work/rest ratios, and discouragement of use of conditioning activities as punishment, but adoption of these policies has been slow.39 In addition, unlike in the military, the athletic guidelines incorporate SCT screening as an adjunct to tailoring precautions, raising concerns about differential treatment of athletes. Encouragingly, however, in recent February 2018 statements, the NCAA reinforced certain preventative measures such as acclimation and work/rest ratios without mention of SCT, reflecting the goal to change culture and incorporate these guidelines for all athletes.40
CKD
SCT has recently been recognized as a risk factor for CKD in both African American and Caribbean populations, conferring a ∼1.5- to 2-fold increased risk of CKD among SCT carriers.41,-43 Unlike exertional injury, CKD is exceedingly common among African American adults; therefore, the overall prevalence of CKD in SCT is relatively high despite the modest relative risk. Specifically, in adults with a mean age of 50 to 65 years, the prevalence of CKD among SCT carriers is ∼15% to 35%, compared with 10% to 25% in individuals without SCT. It is important to note that SCT is therefore a risk factor for CKD, rather than a disease itself, and that several factors are likely at play to result in CKD in the African American population.
Several comorbid conditions are expected to influence CKD among SCT carriers. Type II diabetes, a major risk for CKD, is likely an additive risk factor for CKD, therefore glucose control may decrease CKD incidence in individuals with SCT. Hypertension, which is also thought to be a major risk factor for CKD in African Americans, likely also contributes to CKD risk in SCT; however, interestingly, SCT and hypertension have been found to significantly interact in epidemiologic studies such that the risk of CKD comparing SCT and non-SCT carriers seems to be greater among individuals without hypertension.42 The etiology of this observation is unknown.
Although the pathophysiology of SCT-related nephropathy is not known, it has been hypothesized that subclinical sickling in the hypoxic renal medulla (partial pressure of oxygen, <20 mm Hg) may contribute to vascular disruption, leading to ischemic-reperfusion injury of renal tubules and eventual glomerulosclerosis, as is observed in SCD.41,44 Corroborating histopathologic evidence of tubular and glomerular injuries has been observed in studies of transgenic SCT mice, and postmortem human studies in adults with SCT have shown vasa recta architectural damage45 ; however, the role of sickle hemoglobin (HbS) and erythrocyte rheologic changes as the underlying pathophysiology of these observations has not been proven.
Coinheritance of α-thalassemia mutations offers unique insight into the role of HbS in renal injury in SCT. Polymerization of HbS is highly dependent upon percentage of hemoglobin S. In SCT, HbS concentration per erythrocyte can range from ∼25% to 45%, and studies have shown that coinheritance of α-thalassemia gene deletions decreases the percentage of HbS in a dose-dependent manner.46 This phenomenon has been demonstrated to be clinically relevant in the kidney; in 1 study, increasing copy numbers of the α-thalassemia deletion protected against urinary concentrating defects in individuals with SCT.47 Percentage of HbS also linearly correlated with decreased urinary concentration.
Recently, coinheritance of the common −3.7 kb of α-thalassemia deletion in African Americans was also shown to protect against CKD in a study of ∼2000 SCT carriers.48 Specifically, the risk of CKD among individuals with SCT and no copies of the α-thalassemia deletion was 2.6 (95% confidence interval, 1.7-4.0) compared with 1.1 (95% confidence interval, 0.6-2.3) in SCT carriers with 1 copy of the α-thalassemia mutation. This striking finding suggests that coinheritance of α-thalassemia, which is present in nearly 30% of African Americans with SCT, may nearly eliminate the risk of CKD in SCT. Additional genetic modifiers such as hemoglobin F mutations and HMOX1 may also modify CKD in SCT, but future studies are needed to elucidate these interactions.49
Although the risk of CKD in SCT is well defined, the risk of progression to ESRD remains unclear. A large population study demonstrated a twofold increased risk of progression to ESRD, comparing individuals with SCT with noncarriers,42 but another case-control study among individuals with mostly diabetic or hypertension-attributed nephropathy did not find SCT to be independently associated with ESRD.50 The reasons underlying this discrepancy are not known but may relate to differential risks of progression in the settings of different comorbid conditions. For example, SCT-related renal dysfunction may not result in the same degree of hypertension as other etiologies of CKD, such as APOL1-related nephropathy; therefore, a risk of ESRD in SCT may not be observed in a hypertensive subpopulation.
Because the natural progression of CKD among SCT carriers has not yet been established, and studies of treatments to halt progression of CKD have not been performed, recommendations regarding the need and timing for screening for CKD among SCT carriers do not currently exist. Furthermore, because of the absence of treatment studies in individuals with SCT-related CKD, routine screening for SCT among individuals with CKD is not currently warranted.
RMC
RMC seems to occur nearly exclusively in sickling hemoglobinopathies, namely SCT, based on compelling case series and reports. Given the extreme rarity of this aggressive tumor, prevalence estimates are unknown, and only a minute fraction of individuals with SCT will ultimately develop this complication. Nonetheless, given its overall poor prognosis (95% mortality, particularly with metastatic disease),51 vigilance for this complication among SCT carriers is of paramount importance.
A systematic review of 217 cases of RMC has provided some insights into the presenting symptoms of this disease.52 The median age was found to be 22 years, with nearly 50% occurring in children <21 years of age, and ∼90% of reported cases occurred in individuals with known or later-discovered SCT. Hematuria and flank pain were noted to be the most common presenting signs. On the basis of these descriptions, gross or microscopic hematuria in children and young adults warrants an evaluation with hemoglobin variant analysis (if SCT status is unknown), followed by prompt referral to urology or renal imaging in those found to have SCT. Similarly, although benign hematuria generally seems to be more common among adult SCT carriers based on a single large study,53 children and adults with known SCT who develop new hematuria should undergo prompt urology evaluation, given the poor survival associated with RMC.
Although the pathophysiology of RMC remains unknown, chronic sickling in the hypoxic renal medulla is presumed to play a part. However, additional genetic predispositions, such as in tumor suppressor genes, undoubtedly also contribute to the development of this complication, especially given the younger ages at presentation.51
VTE
Several studies have demonstrated that SCT is a modest risk factor for VTE among African Americans, with an overall 1.5- to 2-fold risk of VTE comparing SCT carriers with noncarriers.54,55 Interestingly, pulmonary embolism rather than deep venous thrombosis seems to nearly fully account for this risk on subgroup analysis, a pattern paradoxical to that observed in general populations but similar to that seen in SCD.56 Although the etiology of this phenomenon is unknown, it has been hypothesized that extreme hypoxia in the venous valves, which can nadir at partial pressure of oxygen <20 mm Hg, as in the renal medulla,57 results in subclinical sickling in SCT, leading to an increased risk of embolization.55 In situ pulmonary artery thrombosis has also been proposed as a mechanism; however, extreme hypoxia to a similar degree does not occur under usual conditions in the pulmonary system but may occur in the setting of underlying parenchymal disease.
The population attributable risk (percentage of VTEs in the population because of SCT) has been calculated as 3% for VTE and 7% for PE,55 which is similar to low-risk thrombophilias such as factor V Leiden and prothrombin gene mutation. Testing for SCT in the setting of VTE is, therefore, likely not warranted, because provoked status is likely to be the major determining factor for recurrence rather than SCT status. Decisions regarding choice and duration of anticoagulation, therefore, should also not be influenced by the presence of SCT. Epidemiologic studies in SCT, however, do offer some clinical insights in monitoring VTE in SCT. Median D-dimer levels are significantly higher in African Americans with SCT compared with noncarriers, with >50% of SCT individuals displaying baseline D-dimers greater than the traditional cutoff for predicting VTE (0.5 µg/mL).58 On the basis of this finding, D-dimer may not be a reliable marker for either ruling out VTE or guiding discontinuation of anticoagulation in individuals with SCT.
Arterial disease
The association of SCT and arterial thrombotic disease has long been speculated, but current evidence is not suggestive of a clear link. The high incidence of childhood stroke in SCD has led to concerns about a similar association in SCT. However, several recent population studies have investigated SCT and ischemic stroke, with the overall evidence demonstrating no association of SCT with incident stroke.59 In addition, a few studies have investigated SCT and cardiac outcomes, although recent evidence suggests no association between SCT and heart failure or risk factors for coronary artery disease, such as hypertension, diabetes, and metabolic syndrome.60,61 Therefore, although it has been hypothesized that the link between SCT and sudden death in the military and athletics may translate into an increased risk of cardiac disease, the current body of literature does not support this assumption. Furthermore, unlike in the renal medulla and venous beds, extreme hypoxia does not generally occur in the arterial system; therefore, pathologic rheologic changes in SCT would not be expected.
Conclusion
SCT is one of the most common hemoglobin carrier states in the world. The expansion of SCT screening and educational efforts, the availability of reproductive technologies, and the increasing research on clinical complications of SCT have important implications for reproductive and genetic counseling guidelines. Future research and clinical efforts are needed to optimize these recommendations for SCT carriers.
Acknowledgments
This work was supported in part by the American Society of Hematology Clinical Scholar Award (L.H.P.) and National Institutes of Health National, Heart, Lung, and Blood Institute grant K08HL125100 (R.P.N.).
Authorship
Contribution: L.H.P. and R.P.N. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence: Rakhi P. Naik, Division of Hematology, Department of Medicine, Johns Hopkins University, 1830 E Monument St, Suite 7300, Baltimore, MD 21287; e-mail: rakhi@jhmi.edu.
