

Key Points
Donor-derived LMP-Ts are safe when administered as adjuvant therapy to prevent relapse after allogeneic HSCT for EBV-associated lymphomas.
Patients had a 2-year OS of 68% that improved to 78% when LMP-Ts were infused in the adjuvant setting.

Abstract
Autologous T cells targeting Epstein-Barr virus (EBV) latent membrane proteins (LMPs) have shown safety and efficacy in the treatment of patients with type 2 latency EBV-associated lymphomas for whom standard therapies have failed, including high-dose chemotherapy followed by autologous stem-cell rescue. However, the safety and efficacy of allogeneic donor-derived LMP-specific T cells (LMP-Ts) have not been established for patients who have undergone allogeneic hematopoietic stem-cell transplantation (HSCT). Therefore, we evaluated the safety and efficacy of donor-derived LMP-Ts in 26 patients who had undergone allogeneic HSCT for EBV-associated natural killer/T-cell or B-cell lymphomas. Seven patients received LMP-Ts as therapy for active disease, and 19 were treated with adjuvant therapy for high-risk disease. There were no immediate infusion-related toxicities, and only 1 dose-limiting toxicity potentially related to T-cell infusion was seen. The 2-year overall survival (OS) was 68%. Additionally, patients who received T-cell therapy while in complete remission after allogeneic HSCT had a 78% OS at 2 years. Patients treated for B-cell disease (n = 10) had a 2-year OS of 80%. Patients with T-cell disease had a 2-year OS of 60%, which suggests an improvement compared with published posttransplantation 2-year OS rates of 30% to 50%. Hence, this study shows that donor-derived LMP-Ts are a safe and effective therapy to prevent relapse after transplantation in patients with B cell– or T cell–derived EBV-associated lymphoma or lymphoproliferative disorder and supports the infusion of LMP-Ts as adjuvant therapy to improve outcomes in the posttransplantation setting. These trials were registered at www.clinicaltrials.gov as #NCT00062868 and #NCT01956084.
Introduction
Although outcomes for most patients with Hodgkin (HL) and non-HL (NHL) are favorable, patients with refractory or relapsed disease have a poor prognosis. Allogeneic hematopoietic stem-cell transplantation (HSCT) may reduce disease relapse compared with autologous HSCT through a graft-versus-lymphoma effect.1,2
Epstein-Barr virus (EBV)–associated lymphomas account for ∼40% of HLs, 20% of diffuse large B-cell lymphomas, and >90% of natural killer (NK)/T-cell lymphomas (NKTCLs),3-5 and immune therapy using EBV-specific T cell–directed therapy has been a successful therapeutic strategy for these patients.6 Although donor lymphocyte infusions (DLIs) may have some efficacy for highly immunogenic type 3 latency tumors, such as posttransplantation lymphoproliferative disorder (PTLD), this approach carries an appreciable risk of graft-versus-host-disease (GVHD) and may be less effective against the less immunogenic type 2 latency lymphomas.7-9
Donor-derived EBV-specific T-cell therapy has proven highly successful in the treatment of PTLD after HSCT, with high efficacy and low rates of GVHD.9-13 Most HLs and NHLs, however, express a more restricted array of EBV antigens (eg, subdominant latent antigens latent membrane protein 1 [LMP1], LMP2, EBNA1, and BARF1)14 and are thus more challenging targets for EBV-specific T-cell therapies. Autologous EBV-specific T cells directed toward LMP1 and LMP2 (LMP-Ts) induced clinical responses in 13 of 21 patients with EBV+ HL and NHL, with a 2-year event-free survival (EFS) of ∼50%, without significant toxicities.6 Seven of 13 patients with B-cell lymphoma and 3 of 8 patients with NKTCL had durable responses.6 Thus, for many patients with relapsed or refractory disease, especially patients with relapsed T cell–derived EBV+ lymphoma or T-cell chronic active EBV (CAEBV), allogeneic HSCT currently offers the only curative approach.15 However, outcomes are typically poor, especially for patients with NK/T-cell disease.15,16,17 Therefore, we evaluated the feasibility, safety, and antitumor activity of donor-derived LMP-T therapy after allogeneic HSCT in patients with EBV+ NK/T-cell or B-cell lymphoma.
Methods
Patients and LMP status of tumors
The protocols for the use of LMP-Ts for patients with EBV+ lymphoma after allogeneic HSCT were approved by the US Food and Drug Administration, US Recombinant DNA Advisory Committee, and Baylor College of Medicine and Children’s National Medical Center institutional review boards and institutional biosafety committees. Informed consent was obtained from patients as well as allogeneic donors.
Twenty-six patients had a diagnosis of EBV+ HL or NHL or EBV-associated) NK/T-cell lymphoproliferative disease, including CAEBV. For these trials, CAEBV was defined as a high EBV viral load in plasma or peripheral blood mononuclear cells (PBMCs; >4000 genomes per microgram of PBMC DNA) and/or biopsy tissue positive for EBV. Immunohistochemistry for LMP1 and/or in situ hydridization for EBER was used to confirm EBV status of tumor biopsies. All patients had undergone allogeneic HSCT. They received LMP-Ts in 2 related phase 1 clinical trials conducted at Baylor College of Medicine or Children’s National Medical Center. Patients with a history of EBV-associated lymphoma or LPD including CAEBV were eligible to receive LMP-T infusions. Patients received LMP-Ts either as treatment of active disease or as adjuvant therapy when the disease was not detectable. All patients received 2 infusions of LMP-Ts 2 weeks apart using a 3 + 3 dose-escalation scheme. The total cell dose ranged from 2 × 107 to 3 × 108 cells per m2 (Tables 1 and 2). Laboratory studies included complete blood counts, comprehensive metabolic panels, and blood for EBV DNA levels and immune reconstitution studies preinfusion and after infusion at 1, 2, 4, and 6 weeks, 3, 6, 9, and 12 months, and yearly for 5 years. Disease reevaluation with imaging was performed at week 8. Patients with active disease at the time of infusion were eligible to receive up to 6 additional infusions if they had stable disease or a partial response at week 8.
Patients in remission at time of LMP-T infusion
| Patient | Institution/ID | Age/sex | Diagnosis | Days post-HSCT | B or T cell | Type of T cell/donor | Total cell dose | Response at 8 weeks postinfusion | Outcome (length of follow-up) | GVHD | Toxicity possibly attributed to LMP-Ts |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | BCM/1145 | 26/M | HL (LMP1+) | 636* | B | LMP2, MRD | 4 × 107/m2 | Relapse | Died of disease (2 mo) | None | None |
| 2 | BCM/1186 | 51/M | DLBCL (EBER+, LMP1+) | 77 | B | LMP2, MUD | 4 × 107/m2 | Remains in remission | Died of HSCT complications (6 mo) | Reactivation; chronic | None |
| 3 | BCM/1242 | 58/M | HL/CLL (EBER+, LMP1+) | 1825* | B | LMP2, MRD | 4 × 107/m2 | Remains in remission | Died of CLL progression (3 y) | None | None |
| 4 | BCM/1285 | 60/F | HL/CLL (LMP1+) | 167 | B | LMP2, MRD | 4 × 107/m2 | Remains in remission | Remains in remission (3+ years) | Chronic | None |
| 5 | BCM/1313 | 44/M | NKTCL (EBER+) | 119 | T | LMP1/2, MRD | 4 × 107/m2 | Remains in remission | Remains in remission (3+ years) | Chronic | None |
| 6 | BCM/1933 | 10/F | CAEBV/HLH (EBV‐ PCR+) | 120 | T | LMP1/2, MUD | 4 × 107/m2 | Remains in remission | Remains in remission† (3+ years) | None | None |
| 7 | BCM/1640 | 13/M | EBV+ LPD (EBER+) | 183 | T | LMP1/2, MRD | 4 × 107/m2 | Remains in remission | Remains in remission (3+ years) | None | None |
| 8 | BCM/1943 | 34/F | HL (EBER+) | 207 | B | LMP1/2, MRD | 1.2 × 108/m2 | Remains in remission | Remains in remission (3+ years) | Reactivation; chronic | None |
| 9 | BCM/2393 | 31/M | NKTCL (EBER+) | 122 | T | LMP1/2, MRD | 1.2 × 108/m2 | Relapse | Died of disease (8 mo) | None | None |
| 10 | BCM/2562 | 17/M | DLBCL (EBER+) | 149 | B | LMP1/2, MUD | 3 × 108/m2 | Remains in remission | Died of disease (3+ years) | Reactivation | None |
| 11 | BCM/2812 | 51/M | DLBCL/CLL (EBER+) | 329 | B | LMP1/2, MRD | 3 × 108/m2 | Remains in remission | Remains in remission (3+ years) | None | None |
| 12 | BCM/2705 | 6/F | HL (EBER+, LMP1+) | 265 | B | LMP1/2, MRD | 3 × 108/m2 | Remains in remission | Remains in remission (3+ years) | None | None |
| 13 | BCM/2879 | 20/F | CAEBV/HLH (EBV‐ PCR+) | 175 | T | LMP1/2, MUD | 3 × 108/m2 | Remains in remission | Remains in remission (3+ years) | None | None |
| 14 | BCM/2997 | 17/F | EBV+ LPD/LYG (EBER+, LMP1+) | 108 | B | LMP1/2, MUD | 4 × 107/m2 (fixed) | Remains in remission | Remains in remission (3+ years) | None | None |
| 15 | BCM/3299 | 12/M | NKTCL (EBER+) | 122 | T | LMP1/2, MRD | 2 × 107/m2 (fixed) | Relapsed | Died of disease (6 wk) | None | None |
| 16 | CNMC/P0148 | 16/M | NKTCL (EBER+) | 42 | T | LMP1/2, MMRD | 2 × 107/m2 | Remains in remission | Remains in remission (13 mo) | None | None |
| 17 | CNMC/P0175 | 22/M | CAEBV (EBV‐PCR+) | 76 | T | LMP1/2, MMRD | 2 × 107/m2 | Relapsed/new NKTCL | Alive with disease (6 mo) | None | None |
| 18 | CNMC/P0182 | 19/M | CAEBV/HLH (EBV‐PCR+) | 84 | T | LMP1/2, MRD | 2 × 107/m2 | Remains in remission | Remains in remission (6 mo) | None | None |
| 19 | CNMC/P0129 | 12/F | NKTCL (EBER+) | 42 | T | LMP1/2, MMRD | 4 × 107/m2 | Remains in remission | Relapsed‡ (15 mo) | None | None |
| Patient | Institution/ID | Age/sex | Diagnosis | Days post-HSCT | B or T cell | Type of T cell/donor | Total cell dose | Response at 8 weeks postinfusion | Outcome (length of follow-up) | GVHD | Toxicity possibly attributed to LMP-Ts |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | BCM/1145 | 26/M | HL (LMP1+) | 636* | B | LMP2, MRD | 4 × 107/m2 | Relapse | Died of disease (2 mo) | None | None |
| 2 | BCM/1186 | 51/M | DLBCL (EBER+, LMP1+) | 77 | B | LMP2, MUD | 4 × 107/m2 | Remains in remission | Died of HSCT complications (6 mo) | Reactivation; chronic | None |
| 3 | BCM/1242 | 58/M | HL/CLL (EBER+, LMP1+) | 1825* | B | LMP2, MRD | 4 × 107/m2 | Remains in remission | Died of CLL progression (3 y) | None | None |
| 4 | BCM/1285 | 60/F | HL/CLL (LMP1+) | 167 | B | LMP2, MRD | 4 × 107/m2 | Remains in remission | Remains in remission (3+ years) | Chronic | None |
| 5 | BCM/1313 | 44/M | NKTCL (EBER+) | 119 | T | LMP1/2, MRD | 4 × 107/m2 | Remains in remission | Remains in remission (3+ years) | Chronic | None |
| 6 | BCM/1933 | 10/F | CAEBV/HLH (EBV‐ PCR+) | 120 | T | LMP1/2, MUD | 4 × 107/m2 | Remains in remission | Remains in remission† (3+ years) | None | None |
| 7 | BCM/1640 | 13/M | EBV+ LPD (EBER+) | 183 | T | LMP1/2, MRD | 4 × 107/m2 | Remains in remission | Remains in remission (3+ years) | None | None |
| 8 | BCM/1943 | 34/F | HL (EBER+) | 207 | B | LMP1/2, MRD | 1.2 × 108/m2 | Remains in remission | Remains in remission (3+ years) | Reactivation; chronic | None |
| 9 | BCM/2393 | 31/M | NKTCL (EBER+) | 122 | T | LMP1/2, MRD | 1.2 × 108/m2 | Relapse | Died of disease (8 mo) | None | None |
| 10 | BCM/2562 | 17/M | DLBCL (EBER+) | 149 | B | LMP1/2, MUD | 3 × 108/m2 | Remains in remission | Died of disease (3+ years) | Reactivation | None |
| 11 | BCM/2812 | 51/M | DLBCL/CLL (EBER+) | 329 | B | LMP1/2, MRD | 3 × 108/m2 | Remains in remission | Remains in remission (3+ years) | None | None |
| 12 | BCM/2705 | 6/F | HL (EBER+, LMP1+) | 265 | B | LMP1/2, MRD | 3 × 108/m2 | Remains in remission | Remains in remission (3+ years) | None | None |
| 13 | BCM/2879 | 20/F | CAEBV/HLH (EBV‐ PCR+) | 175 | T | LMP1/2, MUD | 3 × 108/m2 | Remains in remission | Remains in remission (3+ years) | None | None |
| 14 | BCM/2997 | 17/F | EBV+ LPD/LYG (EBER+, LMP1+) | 108 | B | LMP1/2, MUD | 4 × 107/m2 (fixed) | Remains in remission | Remains in remission (3+ years) | None | None |
| 15 | BCM/3299 | 12/M | NKTCL (EBER+) | 122 | T | LMP1/2, MRD | 2 × 107/m2 (fixed) | Relapsed | Died of disease (6 wk) | None | None |
| 16 | CNMC/P0148 | 16/M | NKTCL (EBER+) | 42 | T | LMP1/2, MMRD | 2 × 107/m2 | Remains in remission | Remains in remission (13 mo) | None | None |
| 17 | CNMC/P0175 | 22/M | CAEBV (EBV‐PCR+) | 76 | T | LMP1/2, MMRD | 2 × 107/m2 | Relapsed/new NKTCL | Alive with disease (6 mo) | None | None |
| 18 | CNMC/P0182 | 19/M | CAEBV/HLH (EBV‐PCR+) | 84 | T | LMP1/2, MRD | 2 × 107/m2 | Remains in remission | Remains in remission (6 mo) | None | None |
| 19 | CNMC/P0129 | 12/F | NKTCL (EBER+) | 42 | T | LMP1/2, MMRD | 4 × 107/m2 | Remains in remission | Relapsed‡ (15 mo) | None | None |
BCM, Baylor College of Medicine; CLL, chronic lymphocytic leukemia; CNMC, Children’s National Medical Center; DLBCL, diffuse large B-cell lymphoma; HLH, hemophagocytic lymphohistiocytosis; LYG, lymphomatoid granulomatosis; MMRD, mismatched unrelated donor; MRD, matched related donor; MUD, matched unrelated donor; PCR, polymerase chain reaction.
Patients had relapsed post-HSCT and attained remission with chemotherapy and/or DLI before receiving LMP-Ts.
Developed a second malignancy (Ewing’s sarcoma) but remains alive and disease free >5 y from LMP-T infusion.
Patient’s donor subsequently diagnosed with NKTCL ∼12 mo after LMP-T infusion.
Patients with refractory or relapsed disease at time of LMP-T infusion
| Patient | Institution/ID | Age/sex | Diagnosis | Days post-HSCT | B or T cell | Type of T cell/donor | Total cell dose | Response at 8 weeks postinfusion | Outcome (length of follow-up) | GVHD | Toxicity possibly attributed to LMP-Ts |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 20 | BCM/2145 | 15/F | CAEBV/HLH (EBV‐PCR+) | 76 | T | LMP1/2, MRD | 1.2 × 108/m2 | PD | Died of disease (2 mo) | None | None |
| 21 | BCM/1898 | 23/F | Hodgkin (EBER+, LMP+) | 1407 | B | LMP1/2, MUD | 2 × 107/m2 (fixed) | SD | Alive with disease (3+ years) | None | None |
| 22 | CNMC/P0085 | 18/F | NKTCL (EBER+) | 111 | T | LMP1/2, MUD | 2 × 107/m2 | PD | Died of disease (3 mo) | None | None |
| 23 | CNMC/P0107 | 10/F | CAEBV (EBV‐PCR+) | 40 | T | LMP1/2, MRD | 2 × 107/m2 | PD | Alive with disease (18 mo) | None | None |
| 24 | CNMC/P0132 | 19/M | HVLL (EBER+) | 63 | T | LMP1/2, MMRD | 4 × 107/m2 | PR | Died of HSCT complications (3 mo) | None | None |
| 25 | CNMC/P0126 | 14/F | CAEBV (EBV‐PCR+) | 35 | T | LMP1/2, MRD | 4 × 107/m2 | PD | Alive with EBV viremia (17 mo) | De novo, grade 1 skin | None |
| 26 | CNMC/P0130 | 2/F | EBV LPD/HLH (EBER+) | 117 | T | LMP1/2, MMRD | 4 × 107/m2 | PR | Died of disease (8 mo) | Reactivation | Grade 4 hepatic necrosis |
| Patient | Institution/ID | Age/sex | Diagnosis | Days post-HSCT | B or T cell | Type of T cell/donor | Total cell dose | Response at 8 weeks postinfusion | Outcome (length of follow-up) | GVHD | Toxicity possibly attributed to LMP-Ts |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 20 | BCM/2145 | 15/F | CAEBV/HLH (EBV‐PCR+) | 76 | T | LMP1/2, MRD | 1.2 × 108/m2 | PD | Died of disease (2 mo) | None | None |
| 21 | BCM/1898 | 23/F | Hodgkin (EBER+, LMP+) | 1407 | B | LMP1/2, MUD | 2 × 107/m2 (fixed) | SD | Alive with disease (3+ years) | None | None |
| 22 | CNMC/P0085 | 18/F | NKTCL (EBER+) | 111 | T | LMP1/2, MUD | 2 × 107/m2 | PD | Died of disease (3 mo) | None | None |
| 23 | CNMC/P0107 | 10/F | CAEBV (EBV‐PCR+) | 40 | T | LMP1/2, MRD | 2 × 107/m2 | PD | Alive with disease (18 mo) | None | None |
| 24 | CNMC/P0132 | 19/M | HVLL (EBER+) | 63 | T | LMP1/2, MMRD | 4 × 107/m2 | PR | Died of HSCT complications (3 mo) | None | None |
| 25 | CNMC/P0126 | 14/F | CAEBV (EBV‐PCR+) | 35 | T | LMP1/2, MRD | 4 × 107/m2 | PD | Alive with EBV viremia (17 mo) | De novo, grade 1 skin | None |
| 26 | CNMC/P0130 | 2/F | EBV LPD/HLH (EBER+) | 117 | T | LMP1/2, MMRD | 4 × 107/m2 | PR | Died of disease (8 mo) | Reactivation | Grade 4 hepatic necrosis |
BCM, Baylor College of Medicine; CNMC, Children’s National Medical Center; HLH, hemophagocytic lymphohistiocytosis; HVLL, hydroa vacciniforme-like lymphoma; MMRD, mismatched unrelated donor; MRD, matched related donor; MUD, matched unrelated donor; PCR, polymerase chain reaction; PD, progressive disease; PR, partial response; SD, stable disease.
Generation of LMP-Ts
We generated good manufacturing practice–compliant LMP-Ts as previously described.18 Briefly, a maximum of 120 mL of whole blood was obtained from healthy stem-cell donors, and PBMCs were separated on lymphocyte separation gradients (Nycomed, Cambridge, MA). PBMCs were used to generate activated monocytes and EBV-transformed B-lymphoblastoid cell lines (LCLs) as antigen-presenting cells, and both were transduced with either an Ad5f35ΔLMP2 vector (n = 4 patients) to overexpress LMP2 or an Ad5f35ΔLMP1-I-LMP2 vector (n = 22) to overexpress LMP1 and LMP2.19-21 For the first stimulation, transduced monocytes were cocultured with donor PBMCs in the presence of interleukin-15 (IL-15; 5 ng/mL; Cellgeneix, Portsmouth, NH). After 10 days, cultures were restimulated with irradiated (50-75 Gy) transduced LCLs in the presence of IL-2 (50-100 units per mL; Prometheus Laboratories Inc., San Diego, CA). Restimulations with transduced LCLs and IL-2 were permitted if cell yield was insufficient. Release testing was performed at the time of cryopreservation of LMP-Ts.
Phenotyping
LMP-Ts were stained with CD3, CD4, CD8, CD16, CD56, CD83, TCRαβ, TCRγδ, CD45, CD19, and CD14 (Miltenyi, San Diego, CA; BD, Franklin Lakes, NJ). For each sample, 2 × 105 cells were analyzed by MACSQuant (Miltenyi) or BD FACSCanto (BD Science, San Jose, CA) using FlowJo software (Treestar, Ashland, OR).
Cytotoxicity assays
A standard 4-hour chromium-51 release assay using effector/target ratios of 40:1, 20:1, 10:1, and 5:1 was used to determine the cytotoxic specificity of each LMP-T product.22 Phytohemagglutinin blasts from either the patient or a related donor were used as target cells.
Enzyme-linked immunospot assay
The LMP1/2 specificity of each LMP-T product was determined by stimulating the cells with EBV pepmixes (JP Peptide Technology, Berlin, Germany) and LCLs and measuring interferon-γ production in enzyme-linked immunospot assays that were read by Zellnet consulting (Fort Lee, NJ). Further characterization to determine epitope specificities was performed using peptide libraries of 15-mer peptides (Genemed Synthesis, San Antonio, TX).
Toxicity monitoring
All patients were monitored for 1 hour after each LMP-T infusion and then for acute GVHD using standard criteria at 2-week intervals for the first 8 weeks23 and for chronic GVHD at 3, 6, 9, and 12 months. This study used the Common Terminology Criteria for Adverse Events of the National Cancer Institute for toxicity reporting.
Statistical analysis
Descriptive statistics were used to summarize spot-forming cells (SFCs) for LMP-Ts at pre- and postinfusion time points as well as the changes in SFCs from preinfusion. Plots of LMP-Ts over time for each patient were generated to visualize patterns of immune reconstitution. Overall survival (OS) was calculated from the time of first LMP-T infusion to death resulting from any cause or censored at last follow-up. EFS was calculated from the time of first LMP-T infusion to the date of first provided relapse, progression, or death or censored at last follow-up. Survival curves were estimated by the Kaplan-Meier method and compared using the log-rank test. P < .05 was considered statistically significantly different.
Data-sharing statement
For original data and protocols, please contact lmclaugh@childrensnational.org. Deidentified individual participant data will not be shared.
Results
Patient characteristics
All patients had undergone allogeneic HSCT for either B- or NK/T-cell EBV-associated lymphoma or LPD (Tables 1 and 2) and received LMP-Ts from the stem-cell donor in 2 related phase 1 clinical trials. A total of 26 patients (median age, 24 years; range, 2-60 years) were available for evaluation (Tables 1 and 2). Twenty-two patients received T cells targeted to LMP1 and LMP2, and 4 received T cells targeted to LMP2. No lymphodepleting chemotherapy was administered before LMP-T infusion. Nineteen patients received LMP-Ts post-HSCT as adjuvant therapy, whereas 7 had active disease at the time of T-cell infusion. Patients who received T cells as adjuvant therapy were treated at a median of 122 days (range, 42-1825 days) post-HSCT.
Characteristics of infused LMP-Ts
LMP-Ts had a median of 55.8% (interquartile range [IQR], 45.2%-70.8%) CD8+ T cells, 8.2% (IQR, 4.5%-28.1%) CD4+ T cells, 11.4% (IQR, 4.4%-16.4%) CD3+/CD56+ NK-like T cells, and 11.2% (IQR, 2.9%-22.4%) CD3−/CD56+ NK cells. No B cells or monocytes were detected in the final product (Figure 1A). No products demonstrated killing of recipient phytohemagglutinin blasts by chromium release assay (data not shown). All products had EBV reactivity to autologous LCLs (median, 160 SFCs per 1 × 105 cells; IQR, 98-295.5 cells), and most had LMP2-specific activity (median, 161.5 SFCs per 1 × 105 cells; IQR, 53.5-356.5) and/or LMP1-specific activity (median, 10 SFCs per 1 × 105 cells; IQR, 2-63.5), as determined in interferon-γ enzyme-linked immunospot assays (Figure 1B). Most products recognized at least 2 LMP2 epitopes restricted by >1 HLA allele (Figure 1C-D).

Characteristics of LMP-T products. (A) Immunophenotyping at time of cryopreservation showed a predominance of CD3+ and CD8+ T cells. Monocytes and B cells were not present in the final products. (B) A majority of products demonstrated robust LMP2 activity, and all products had EBV activity, as demonstrated by their response to LCLs in interferon-γ enzyme-linked immunospot assays. (C-D) Number of LMP1 and LMP2 epitopes and HLA alleles recognized by LMP-specific product. Although we were not able to identify a specific LMP1 epitope recognized by most products, a majority of products recognized ≥2 LMP2 epitopes and HLA alleles.
Characteristics of LMP-T products. (A) Immunophenotyping at time of cryopreservation showed a predominance of CD3+ and CD8+ T cells. Monocytes and B cells were not present in the final products. (B) A majority of products demonstrated robust LMP2 activity, and all products had EBV activity, as demonstrated by their response to LCLs in interferon-γ enzyme-linked immunospot assays. (C-D) Number of LMP1 and LMP2 epitopes and HLA alleles recognized by LMP-specific product. Although we were not able to identify a specific LMP1 epitope recognized by most products, a majority of products recognized ≥2 LMP2 epitopes and HLA alleles.
Safety and toxicity
There were no immediate infusion-related toxicities or adverse events. Only 1 dose-limiting toxicity was considered probably related to T-cell infusions in patient 26, who received parental donor-derived LMP1/2-specific T cells on day +117 after haploidentical HSCT for a T cell–associated EBV LPD with EBV-associated HLH. At the time of T-cell infusion, 16 810 EBV DNA copies per mL of PB were detected, but her HLH remained quiescent. She tolerated LMP-T infusion well, but 2 days after the second infusion and day +16 after the first (total dose, 4 × 107cells per m2), she developed intermittent fevers, raising concern of progressive disease. A positron emission tomography (PET)/computed tomography scan showed new [18F]fluorodeoxyglucose-avid lesions in the mediastinum, liver, kidney, and abdomen, determined to be due to disseminated Mycobacterium avium complex. However, liver biopsy also showed grade 4 necrosis, but with small focal collections of EBER+ cells localized to the edges of the necrotic tissue, together with CD3+ T cells (Figure 2). Despite hepatic necrosis, the patient did not have significant changes in hepatic function (aspartate aminotransferase, 151 U/L; alanine aminotransferase, 105 U/L; total bilirubin, 0.3 mg/dL). The patient subsequently received third-party EBV-specific T cells ∼3 months after LMP-T infusion because of rising EBV viremia, but with poor response, and ultimately relapsed with HLH ∼5 months after LMP-T infusion. She underwent HLH induction therapy as a bridge to a second allogeneic HSCT but ultimately died 2 months posttransplantation.
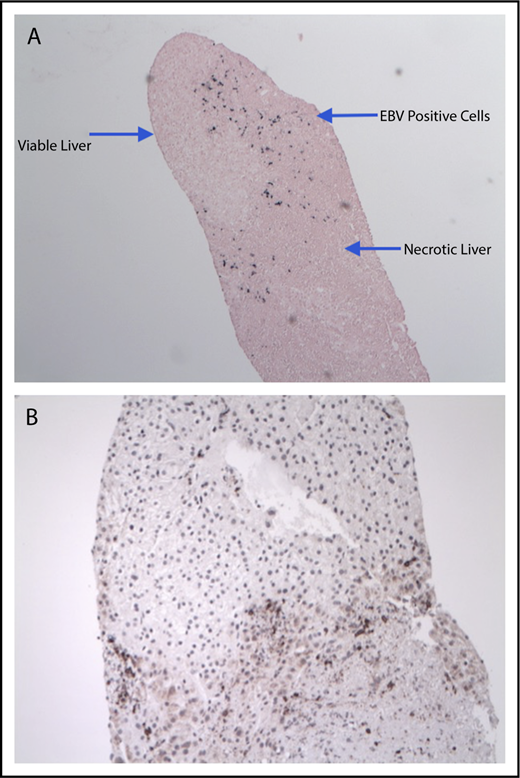
Hepatic necrosis in a patient after receiving LMP-T infusion. EBER in situ hybridization of liver biopsy showed EBV+ cells along the borders of necrotic hepatic tissue (original magnification ×4) (A), and CD3 immunohistochemical staining reveled numerous T lymphocytes in the same region (original magnification ×10) (B).
Hepatic necrosis in a patient after receiving LMP-T infusion. EBER in situ hybridization of liver biopsy showed EBV+ cells along the borders of necrotic hepatic tissue (original magnification ×4) (A), and CD3 immunohistochemical staining reveled numerous T lymphocytes in the same region (original magnification ×10) (B).
One patient (25) developed de novo grade 1 acute skin acute GVHD on day +6 after the first LMP-T infusion (day +41 from a her matched-sibling PBSC HSCT), when calcineurin inhibitor levels were found to be subtherapeutic. The rash resolved within 2 days of topical steroid therapy, and a second LMP-T infusion was administered without GVHD recurrence. Two additional patients with a history of acute GVHD before LMP-T infusion developed late acute GVHD after infusion. Patient 10 had a history of grade 2 gut GVHD and developed grade 2 acute skin GVHD on day +83 after LMP-T infusion, but this resolved after treatment with steroids. Patient 2 had a history of acute skin GVHD that reactivated (initially grade 1) 2 months after LMP-T infusion. He ultimately progressed with gut and liver involvement and required systemic therapy, which likely contributed to subsequent reactivation of BK viremia. The patient died 6 months after study entry from sepsis resulting from GVHD and increased immunosuppression. Three additional patients (patients 4, 5, and 8) developed limited chronic GVHD, including 1 who had a reactivation of acute skin GVHD.
Outcomes after LMP-T infusion
The 2-year EFS and OS rates for all 26 patients were 46% and 68%, respectively (Figure 3A-B). In our cohort, patients with B-cell disease (n = 10) had better outcomes than those with T cell–mediated disease (n = 16), with an 80% 2-year OS compared with a 60% 2-year OS (Figure 3C). These data compare favorably with expected outcomes based on Center for International Blood and Marrow Transplant Research (CIBMTR) data for patients with NK/T cell–mediated disease (2-year OS, 36%) and those with B cell–derived lymphoma (2-year OS, ∼60%-75%).17,24 In this current study, there was a trend of responders receiving T-cell products containing higher specificity for LMP2/EBV antigens (Figure 4; not statistically significant). Furthermore, as with patients receiving autologous LMP-Ts, there was a trend of responding patients having higher frequencies of circulating LMP2-specific T cells detected in the PB compared with nonresponders (Figure 5; not statistically significant).6

Outcomes in patients who received LMP-T products. (A) Two-year EFS. (B) Two-year OS. (C) Patients with B cell–mediated disease had overall improved survival compared with patients with T cell–mediated disease.
Outcomes in patients who received LMP-T products. (A) Two-year EFS. (B) Two-year OS. (C) Patients with B cell–mediated disease had overall improved survival compared with patients with T cell–mediated disease.

LMP-specific activity of LMP-T products. Responders (Rs) received LMP-T products with slightly higher LMP2-specific and EBV activity (as demonstrated by using LCLs as stimulators in interferon-γ enzyme-linked immunospot assays, which may overlap with LMP1 and/or 2 responses) compared with nonresponders (NRs).
LMP-specific activity of LMP-T products. Responders (Rs) received LMP-T products with slightly higher LMP2-specific and EBV activity (as demonstrated by using LCLs as stimulators in interferon-γ enzyme-linked immunospot assays, which may overlap with LMP1 and/or 2 responses) compared with nonresponders (NRs).

Frequency of LMP2-specific T cells in responding vs nonresponding patients. PB samples at set time points after T-cell infusion were incubated with LMP2 pepmixes and then plated in interferon-γ enzyme-linked immunospot assays. Responding patients (A) had a slightly greater frequency of LMP2-specific T cells than nonresponders (B).
Frequency of LMP2-specific T cells in responding vs nonresponding patients. PB samples at set time points after T-cell infusion were incubated with LMP2 pepmixes and then plated in interferon-γ enzyme-linked immunospot assays. Responding patients (A) had a slightly greater frequency of LMP2-specific T cells than nonresponders (B).
Impact of disease status at time of T-cell infusion on outcome
Patients in remission
Patients who received T-cell therapy while in complete remission after allogeneic HSCT had a 57% 2-year EFS and a 78% 2-year OS. This is compared with the published EFS of 30% to 50% and OS of 36% to 75% in T-cell and B-cell lymphoma patients after allogeneic HSCT.2,17,24,25 In the current study, 2 patients with CAEBV developed new malignancies after LMP-T therapies. Patient 6 developed Ewing’s sarcoma (22 months after HSCT and 18 months LMP-T infusion), which was successfully treated, and the patient remains in long-term remission. Patient 17 underwent HSCT for T-cell CAEBV and developed a nasal NKTCL 6 weeks after LMP-T infusion. He is currently undergoing chemotherapy and radiation therapy. In retrospect, it is suspected that this patient had NKTCL that was undiagnosed pretransplantation, because he did not have imaging of the nasal area at that time.
Five patients died as a result of their underlying CAEBV/EBV-associated lymphoma. Patient 9 had a relapse of his NKTCL and died 8 months after receiving LMP-Ts, and patient 15 with NKTCL relapsed rapidly and died within 6 weeks of receiving LMP-Ts. Patient 10 had a very late relapse of his EBV+ diffuse large B-cell lymphoma >3 years after HSCT and LMP-T infusions and subsequently died as a result of his lymphoma. Patient 3 had a history of chronic lymphocytic leukemia that transformed into EBV+ HL (Richter’s syndrome). Although he remained in long-term remission from his EBV+ HL, he died as a result of chronic lymphocytic leukemia progression ∼3 years after LMP-T infusion. Patient 1 initially was diagnosed with EBV+ HL and received 2 doses of LMP-specific T cells as adjuvant therapy. Although a PET scan 10 days before his first dose of LMP2-specific T cells showed no evidence of active disease, he developed enlarged left supraclavicular and axillary lymph nodes, and lymph node biopsy showed relapse with EBV− HL 26 days after the second LMP-T infusion (Figure 6). He subsequently died as a result of disease progression.

Lymphoma relapse with loss of EBV positivity. (A) A patient who received LMP2-specific T cells as adjuvant therapy after undergoing allogeneic transplantation for an EBV+ HL, as demonstrated by positive LMP1 staining (×40), was noted to have progressive disease shortly after receiving LMP2-specific T cells. (B) However, biopsy of the relapsed lymphoma demonstrated that the tumor was no longer EBV+.
Lymphoma relapse with loss of EBV positivity. (A) A patient who received LMP2-specific T cells as adjuvant therapy after undergoing allogeneic transplantation for an EBV+ HL, as demonstrated by positive LMP1 staining (×40), was noted to have progressive disease shortly after receiving LMP2-specific T cells. (B) However, biopsy of the relapsed lymphoma demonstrated that the tumor was no longer EBV+.
Patients with relapsed disease at the time of infusion
Patients with lymphoma (especially T- and B-cell NHL) who have already relapsed after allogeneic HSCT have an extremely poor prognosis, with an expected EFS and OS of <20% at 2 years.25-27 In our cohort, the 7 patients who received LMP-Ts for persistent or relapsed disease after HSCT had an OS of 43% at 2 years (Figure 7A-B). Two patients had a partial response to LMP-T infusion. Patient 24 with hydroa vacciniforme-like lymphoma had a very good partial response, with EBV viremia decreasing from 464 482 DNA copies per mL at the time of first infusion to 34 965 DNA copies per mL, but the patient died as a result of complications related to HSCT (pulmonary venoocclusive disease and thrombotic microangiopathy) 4 months after LMP-T infusion. Patient 26 with EBV HLH/LPD had a partial response to LMP-Ts, which was based on the detection of areas of necrosis in EBV+ hepatic tissue. However, the patient subsequently had a rising EBV viral load and a flare of HLH. She received third-party EBV-specific T cells ∼3 months after receiving LMP-Ts and proceeded to a second HSCT but ultimately died as a result of disease progression. Patients 20 and 22 with T cell–mediated disease (CAEBV/HLH and NKTCL) developed disease progression within 1 month of LMP-T infusion and died as a result of progressive disease within 3 months of infusion. Despite relapsing after donor-derived LMP-Ts, 2 patients were salvaged with additional immunotherapy with DLI, suggesting that broadening the antigen-specific activity of the infused T-cell product may be important. This is evidenced by the fact that 2 patients with CAEBV without a history of lymphoma (patients 23 and 25) developed progressive disease (as evidenced by elevated EBV DNA levels) after LMP-T infusion and subsequently received DLIs from the same donor. Patient 23 received a DLI without lymphodepleting chemotherapy and continued to have high levels of EBV viremia but clinically is well and asymptomatic. Patient 25 received lymphodepleting chemotherapy before DLI and obtained a complete response with undetectable EBV DNA levels, which she maintained for ∼12 months, at which time low levels of EBV viremia were detected.

Outcomes for patients who received LMP-Ts as adjuvant therapy vs treatment for active disease. Patients who received LMP-Ts as adjuvant therapy after HSCT had superior 2-year EFS (A) and OS (B) compared with patients who had active disease at the time of LMP-T infusion.
Outcomes for patients who received LMP-Ts as adjuvant therapy vs treatment for active disease. Patients who received LMP-Ts as adjuvant therapy after HSCT had superior 2-year EFS (A) and OS (B) compared with patients who had active disease at the time of LMP-T infusion.
Discussion
We have shown that donor-derived LMP-Ts can safely maintain clinical responses when administered as adjuvant therapy to patients with high-risk EBV-related lymphoma or lymphoproliferative conditions after allogeneic HSCT. No patient had any immediate adverse events related to infusion. The only patient who developed de novo GVHD was undergoing subtherapeutic immune suppression when GVHD developed and responded quickly to treatment with topical steroids. Only 1 dose-limiting toxicity occurred but without significant clinical impact on the patient or her long-term outcome. Although patients had overall 2-year EFS and OS rates of 46% and 68%, respectively, patients in complete remission who received LMP-Ts as adjuvant therapy after allogeneic HSCT had a 57% 2-year EFS and a 78% 2-year OS despite having aggressive or relapsed or refractory disease. Hence, we conclude that LMP-directed T-cell therapy may be beneficial when administered as adjuvant therapy after HSCT before disease is detected by imaging and/or EBV polymerase chain reaction assays.
Despite overall improved outcomes for patients with newly diagnosed HL or NHL, those with refractory or relapsed disease or T- or NK-cell disease generally fare worse. For these patients, allogeneic HSCT with reduced-intensity regimens to decrease nonrelapse mortality can be effective.28 Although chronic GVHD has been associated with a decreased risk of relapse in hematologic malignancies, presumably because of an overlap with a potent graft-versus-tumor effect, the increased risk of transplantation-related mortality in patients with chronic GVHD results in worse OS.1,29 Therefore, we proposed the use of EBV-specific LMP-Ts to bolster the graft-versus-tumor effect while minimizing the risk of GVHD compared with DLI. LMP1- and LMP2-specific T cells have previously been generated from EBV+ lymphoma patients and can induce or maintain durable complete responses in patients with high-risk disease, which supports the use of LMP 1 and LMP2 as suitable targets for immune therapies directed against EBV-associated lymphoma.6,30,31 Here we show that LMP-Ts derived from healthy donors can be safely infused and may prevent relapse when administered as adjuvant therapy to high-risk patients after allogeneic HSCT.
Although the safety of an allogeneic T-cell product was the primary end point of these trials, efficacy was also evaluated. Unfortunately, there is no direct historical cohort for comparison, but based on CIBMTR data, the 2-year EFS after allogeneic HSCT for diffuse large B-cell NHL (EBV status undetermined) is ∼50% in the pediatric setting.25 In patients with T-cell lymphoma, the 2- to 5-year OS ranges from 30% to 50%.16 In the largest CIBMTR analysis of patients with extranodal NKTCL undergoing allogeneic transplantation, the 3-year progression-free survival and OS rates were only 28% and 34%, respectively.17 In contrast, the 2-year OS in our cohort was 78% when patients received LMP-Ts as adjuvant therapy. Patients with B-cell disease had better outcomes than those with T cell–mediated disease (2-year OS, 80% vs 60%); because B cells are professional antigen-presenting cells, we posit that an EBV+ B-cell malignancy would be more responsive to EBV-directed T-cell therapeutics compared with an EBV+ T-cell malignancy. However, the 2-year OS of 60% in our patients with T-cell disease seems superior to that in historical cohorts16,17 and includes 2 patients (patients 5 and 16) with aggressive EBV+ NKTCL who remain alive and disease free for 1 to 3+ years after receiving donor-derived LMP-Ts. These data suggest that LMP-Ts may be an effective therapy to prevent relapse, given the unfavorable outcome in patients with highly aggressive and refractory disease, especially those with T cell–mediated disease.16,17,25,26
Although LMP-T therapy seemed effective in maintaining remission after allogeneic HSCT, it was unable to induce complete remission in all patients with relapsed disease after allogeneic HSCT. Two patients with aggressive T-cell disease achieved a disease response with LMP-Ts alone. However, despite relapsing after donor-derived LMP-Ts alone, 2 additional patients were salvaged with DLI, suggesting that broadening the antigen-specific activity of the infused T-cell product may be important for the future development of EBV antigen–directed T cell–based therapeutics for this poor-prognosis disease as well as comprehensive evaluation of EBV antigen expression in tumor samples obtained preinfusion. Such an approach has already been developed for the autologous setting, where next-generation T-cell therapies for type 2 latency EBV+ lymphoma also target EBNA1 and BARF1 in addition to LMP1 and LMP2.32,33
With any targeted therapy, there is a concern about antigen loss, as suggested by the patient who relapsed with EBV− HL after LMP-T infusion. However, because he was diagnosed with EBV− HL only 26 days after receiving the second LMP-T infusion, we posit that despite a negative PET scan pre–T-cell infusion, it is likely that the patient had subclinical disease present at the time that was already EBV−. Thus, his rapid progression and death shortly after receiving LMP-T therapy were less likely a failure of the treatment, but they emphasize the importance of obtaining biopsies to determine EBV status at the time of relapse.
A practical limitation to our prophylactic treatment strategy was the availability of LMP-T products, given the time needed to manufacture the products and the inability of patients to travel to the clinical site to receive T-cell infusions early after HSCT, while still in remission, because of transfusion dependence, infections, and poor nutritional status. Additionally, common post-HSCT complications such as GVHD and abnormal renal and hepatic function affected patients’ eligibility for the clinical trial and therefore contributed to delays in the administration of LMP-Ts post-HSCT. Given the superior results using LMP-Ts as adjuvant therapy, we recommend administering LMP-Ts as soon as possible after day +30 when the donor graft has become established. This requires early referral and coordination of care. Another concern is that a related donor may also have an underlying genetic predisposition to T cell–mediated EBV malignancies. Evidence for this concern was highlighted by patient 20 with NKTCL, whose donor (mother) had no evidence of underlying EBV immune dysfunction at the time of selection as the HSCT and LMP-T donor. However, the mother developed an EBV-associated NKTCL approximately 1 year after her daughter underwent the HSCT followed by LMP-T infusion. The daughter subsequently relapsed 16 months post–LMP-T infusion (17.5 months post-HSCT). To circumvent this potential complication, future studies may also consider the use of third-party off-the-shelf EBV T cells as prophylaxis in this high-risk setting, as is currently being evaluated for patients with PTLD after solid organ transplantation (trials registered at www.clinicaltrials.gov as #NCT02900976 and #NCT03394365) or HSCT (#NCT03392142). However, unlike donor-derived patient-specific EBV T-cell products, which have persisted up to 10 years when used to prevent PTLD post-HSCT, the use of third-party products does not necessarily restore long-term immune reconstitution.13,34
In conclusion, we have shown that donor-derived LMP-Ts can safely be infused into patients with EBV-associated lymphoma or LPD after undergoing allogeneic HSCT, including patients with relapsed T cell–mediated disease, who generally have extremely poor outcomes in this setting. Although patients with T-cell disease had worse outcomes than patients with B-cell disease, our results seem better than historical outcomes and therefore support the administration of EBV-directed T-cell therapy in the adjuvant setting as a means of preventing relapse in these highest-risk patient populations.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health (NIH), National Cancer Institute grant PO1 CA094237, Lymphoma SPORE grant P50 CA126752, and Leukemia and Lymphoma Society SCOR grant 7018-04 (H.E.H.); Children’s Oncology Group grant FP00015221_SUB706_01 (C.M.B.); a Lymphoma Research Foundation Career Development Award (L.P.M.); St Baldrick’s grant 300001991 48-051-3 (C.M.B.); shared resources of the Dan L. Duncan Comprehensive Cancer Center support grant P30CA125123; and NIH National Center for Advancing Translational Sciences award number UL1TR001876.
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Advancing Translational Sciences or the NIH.
Authorship
Contribution: L.P.M. and R.R. contributed to data collection and interpretation and writing of the manuscript; M.K.B., C.M.R., H.E.H., and C.M.B. contributed to the study conception and design, data collection and interpretation, and writing of the manuscript; M.-F.W. contributed to data interpretation and writing of the manuscript; V.T., F.H., and B.G. contributed to data collection; A.M.M. and G.C. contributed to data collection and interpretation; S.G. contributed to study conception and design and developed the Ad5f35LMP1-I-2 vector; P.J.H. and A.P.G. contributed to the study design and manufacturing of products; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: C.M.B. is on the scientific advisory boards of Cellectis, Torque, and Neximmune and has stock or ownership in Mana Therapeutics; S.G. has a consulting or advisory role with AbbVie, Celgene, and Merrimack Pharmaceuticals and several patent applications (8 focused on cell therapy for cancer, 1 focused on oncolytic viruses, and 1 focused on cancer gene therapy); M.K.B. has stock or ownership in Viracyte and Marker Therapeutics, has a consulting or advisory role with Torque, Unum, and Tessa Therapeutics, and receives research funding from Cell Medica and Tessa Therapeutics; H.E.H. has stock or ownership in Viracyte and Marker Therapeutics, has a consulting or advisory role with Gilead and Novartis, receives research funding from Cell Medica and Tessa Therapeutics, and has patents/royalties/other intellectual property through Cell Medica; C.M.R. has stock or ownership in Viracyte and Marker Therapeutics, has a consulting or advisory role with Cell Medica, CellGenix, and Tessa Therapeutics, receives research support from Tessa Therapeutics, and has a patent application in the field of cellular immunotherapy; and R.R. has received honoraria for serving on a Novartis Treatment Advisory Landscape Advisory Board regarding CAR T-cell commercialization. The remaining authors declare no competing financial interests.
Correspondence: Catherine M. Bollard, Center for Cancer and Immunology Research, George Washington University, 111 Michigan Ave NW, Washington, DC 20010; e-mail: cbollard@childrensnational.org.
