

In this issue of Blood, 1 shed light on the mechanism by which the deubiquitinating hydrolase, UCH1L, promotes MYC-driven neoplasms of mature B cells and plasma cells. The new findings solve the long-standing conundrum of UCH1L’s ability to maintain high-level protein synthesis despite inhibiting its positive upstream regulator, mTORC1.
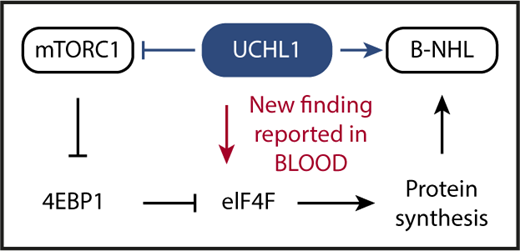
Schematic depiction of the key advance reported by Hussain et al in this issue of Blood. UCHL1 is a deubiquitinating enzyme that has been shown in previous work to promote malignant B-cell and plasma cell development (indicated by blue arrow pointing right) despite inhibiting mTORC1 (blue horizontal line with a short vertical “stop” line). The inhibition of mTORC1 has been enigmatic because the protein complex serves as a crucial positive regulator of cap-dependent mRNA translation and protein synthesis: a mandatory requirement for malignant growth. Hussain et al solved the enigma by demonstrating that UCHL1 promotes the assembly of the eIF4F translation initiation complex (red arrow pointing down), bypassing, thereby, the inhibitory impact of the UCHL1-mTORC1-4EBP1 axis on protein synthesis. Hussain et al also show that the enzymatic, deubiquitinating activity of UCHL1 is crucial for MYC-driven lymphoma in laboratory mice. This provided preclinical support for the contention that targeted inhibition of deubiquitination using small-molecule drugs may lead to new treatments for patients with lymphoma and myeloma.
Schematic depiction of the key advance reported by Hussain et al in this issue of Blood. UCHL1 is a deubiquitinating enzyme that has been shown in previous work to promote malignant B-cell and plasma cell development (indicated by blue arrow pointing right) despite inhibiting mTORC1 (blue horizontal line with a short vertical “stop” line). The inhibition of mTORC1 has been enigmatic because the protein complex serves as a crucial positive regulator of cap-dependent mRNA translation and protein synthesis: a mandatory requirement for malignant growth. Hussain et al solved the enigma by demonstrating that UCHL1 promotes the assembly of the eIF4F translation initiation complex (red arrow pointing down), bypassing, thereby, the inhibitory impact of the UCHL1-mTORC1-4EBP1 axis on protein synthesis. Hussain et al also show that the enzymatic, deubiquitinating activity of UCHL1 is crucial for MYC-driven lymphoma in laboratory mice. This provided preclinical support for the contention that targeted inhibition of deubiquitination using small-molecule drugs may lead to new treatments for patients with lymphoma and myeloma.
Ubiquitination is a sequential enzymatic process that covalently attaches the 76-residue polypeptide ubiquitin to client proteins. This either targets them for proteasomal degradation or regulates their functional properties, such as enzymatic activity, subcellular localization, and interaction with other proteins in supramolecular complexes. Just like other posttranslational protein modifications, ubiquitination is reversible: a job performed by a sizable family of peptidases called deubiquitinases (n ≤ 100). These can be classified into 6 subfamilies based on sequence and domain conservation, and are able to cleave ubiquitin from target proteins, edit ubiquitin chains on proteins, or process ubiquitin precursors in order to maintain a pool of free ubiquitin necessary for normal cell function. Ubiquitin C-terminal hydrolase L1, or UCHL1 for short, is the founding member of the deubiquitinase family, which is currently emerging as a promising molecular target for new approaches to cancer therapy and prevention.2
Previous studies by the investigator team reporting in Hussain et al, ingeniously led for more than a decade by Paul Galardy at Mayo Rochester, established that upregulation of UCHL1 prognosticates poor outcome of patients with diffuse large B-cell lymphoma3 and multiple myeloma.4 Consistent with an oncogenic role in mature B cells and plasma cells, transgenic expression of UCHL1 accelerated neoplastic B-cell development in laboratory mice,5 including MYC-driven lymphoma in strain EμMyc,6 a widely used model of deregulated MYC protooncogene expression consequent to human Burkitt lymphoma t(8;14)(q24;q32) translocation. Interrogation of the mechanism by which UCHL1 promotes malignant B cell and plasma cell development demonstrated that UCHL1 impacts a cellular signal transduction module that is widely considered the master regulator of cell growth: mammalian (mechanistic) target of rapamycin, mTOR.7
mTOR is a serine/threonine kinase that, by virtue of assembling with different scaffold proteins and cofactors, forms 2 principal complexes with distinct biological functions: raptor-bound mTOR, designated mTORC1, governs 5′-terminal cap (7-methylguanosine, m7G)-dependent messenger RNA (mRNA) translation, whereas rictor-bound mTOR, dubbed mTORC2, activates a downstream serine/threonine kinase that is crucial for cell proliferation and survival: AKT (v-Akt murine thymoma viral oncogene homolog). Interestingly, Galardy and associates showed in earlier work that in B lymphocytes UCHL1 shifts the balance of mTORC1 to mTORC2 activity in favor of the latter. This relies on a mechanism that involves the deubiquitination of the scaffold protein, raptor, and a loss of mTORC1 kinase activity that results, in turn, in reduced phosphorylation-dependent inhibition of downstream substrates such as 4EBP1, which is eukaryotic translation initiation factor 4E-binding protein 1. 4EBP1 is a negative regulator of protein synthesis that inhibits the eukaryotic translation initiation factor 4F complex, eIF4F, that is key for cap-dependent translation and protein synthesis. Increased levels of 4EBP1, caused by UCHL1-dependent suppression of mTORC1, are expected to result in strong eIF4F inhibition and low protein synthesis (see figure).
Although the above-mentioned UCHL1-dependent increase in mTORC2 activity leading to enhanced AKT survival and proliferative signaling provides an obvious advantage for neoplastic development, the inhibition of mTORC1 by UCHL1 presented a conundrum given the importance of protein biosynthesis in cancer in general and in MYC-driven lymphoma in particular. To attack this problem and further our understanding of UCHL1’s oncogenic function, Hussain et al took advantage of proximity-based proteomics to demonstrate that UCHL1 is associated with and promotes the assembly of the eIF4F translation initiation complex. The experimental results show that UCHL1, by virtue of bypassing 4EBP1 and targeting eIF4F directly, is able to drive protein biosynthesis despite suppressing mTORC1 (see figure, red arrow). Another significant advance made by Hussain et al relied on a newly developed mouse model of transgenic expression of hydrolase-dead UCHL1, demonstrating that the enzymatic deubiquitinating activity of the protein is important for MYC-driven lymphoma.
In summary, Hussain et al identified a novel mechanism that permits UCHL1 to drive protein biosynthesis in the face of mTORC1 inhibition. In addition, the authors demonstrate an important role of deubiquitination in MYC-driven lymphoma development. The new results lend strong preclinical support to the design and testing of small-compound inhibitors that target UCHL1 and related deubiquitinating enzymes for therapeutic purposes.8 In myeloma, for example, inhibitors of this sort may overcome acquired resistance to proteasome inhibition.9 This was recently shown for PSMD14 (proteasome 26S subunit, non-ATPase 14), a deubiquitinase that resides in the 11S cap of the 20S proteasome.10 For myeloma and lymphoma patients who are relapsed or refractory to standard therapies, deubiquitinase inhibitors cannot come down the road soon enough.
Conflict-of-interest disclosure: S.J. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal