

Abstract
Introduction
von Willebrand disease (VWD) is a common inherited bleeding disorder with a reported incidence ranging from 0.01% to 1%. VWD is classified as quantitative (types 1 and 3) and qualitative (type 2) defects in von Willebrand factor (VWF). Classical VWD type 2A, the most common qualitative defect of VWD, is characterized by decreased VWF-dependent platelet adhesion and a selective deficiency of high molecular weight VWF multimers. The majority of type 2A VWD is inherited in an autosomal dominant fashion. This subtype is caused by missense variants acting through a variety of mechanisms including defects in multimer assembly or an intrinsically increased sensitivity to cleavage by ADAMTS-13. The majority of pathogenic mutations are located in exon 28, predominantly affecting the A2 domain. Missense mutations have also been reported in the D3 and A1 domains (exons 22 and 25-28). Less common missense mutations include those responsible for dimerization (CK domain, exon 52) and multimerization defects (D2 domain, exons 11-17). Our objective was to determine the genomic distribution of mutations underlying type 2A VWD in patients referred to a USA-based clinical reference laboratory.
Methods
In a clinical reference laboratory, BloodCenter of Wisconsin, VWF gene sequence analysis results from January 2009 to July 2018 were reviewed. Molecular testing was performed by polymerase chain reaction (PCR) amplification and bi-directional Sanger sequencing or capture-based enrichment and next-generation sequencing (NGS). Complete coding region and splice junction of VWF gene was compared to the reference sequence (NM_00552.3). Variant interpretation was performed according to American College of Medical Genetics (ACMG) standards and guidelines by utilizing genome databases, population frequency data, variant specific literature search and predictive computational tools. Variant interpretation was reviewed by a team comprising of genetic counselors, molecular experts and experts in coagulation disorders correlating these findings with the patient's clinical phenotype provided.
Results
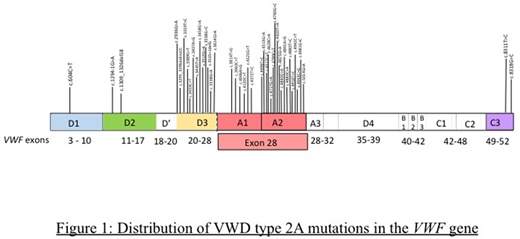
Pathogenic variants were identified in 46 cases received. Of these, 56.5 % (26 of 46) were in exon 28 of the VWF gene.(Figure 1) 21.7% of mutations (10 out of 46) were identified in exon 26, 4.3% were identified in exon 25 (2 of 46) and 6.5% (3 out of 46) were identified in exon 52 of VWF gene. The remaining 10.7% were identified in exons 6,11,12,22 and 27 (1 each). The distribution of these mutations according to VWF domains was also studied. 39% of these mutations were found in the A2 domain, 30.4% were identified in the D3 domain, and 13% were found in the A1 domain. A small number of these mutations were identified in the D1 domain (1/46), D2 domain (2/46) and CK domain (2/46). Phenotypic information was available for most patients evaluated.
Conclusions
In a predominantly Caucasian population with others of mixed ethnicities, the distribution of VWD type 2A mutations was studied in patients evaluated in a clinical reference laboratory. Most mutations were identified in VWF Exon 28 and in the A2 domain. Next generation sequencing platform enables sequencing of large amounts of DNA in parallel thereby allowing all VWF exons to be evaluated at one time. However, in smaller laboratories, without access to a NGS platform, initial evaluation of the VWF Exon 28 for evaluation of VWD type 2A variants is a cost effective measure. Subsequently, sequencing of other less implicated exons may be performed utilizing a reflexive algorithm. Genetic testing can be very useful in diagnosis of type 2 variants, where mutations in specific regions correspond to the defect in VWF function.
Friedman:Shire: Membership on an entity's Board of Directors or advisory committees; CSL Behring: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal