

Abstract
Introduction: Relapse of AML after initial remission occurs in about 30-50% of patients (pts) and is associated with dismal outcome. The majority of pts relapse within the first 2 years after diagnosis and their clonal evolution has been extensively studied. Late relapses, however, are very rare and therefore poorly characterized. We set out to study pts who experience relapse >3 years after initial diagnosis to depict the genetic profile and gain knowledge about the pathogenesis in these pts.
Methods: We selected AML cases sent to our laboratory for diagnostic work-up from 2005 - 2015 and who relapsed > 3 years after initial diagnosis (Dx). Based on sample availability, whole exome sequencing (WES) was carried out for 31 pts at the time points of Dx and relapse as well as on matched remission samples that were available for 19 pts. Enrichment based library preparation was performed using the xGen Exome Research Panel and sequenced (2x151bp) on a NovaSeq instrument. Data was processed with BaseSpace using the BWA Enrichment app with BWA for Alignment (against hg19) and GATK for variant calling with default parameters. Data was subsequently loaded into BaseSpace Variant Interpreter to filter and prioritize variants of interest. Only passed protein changing variants were considered with an ExAC population frequency of less than 1% for further analysis. As control cohort, we identified patients that relapsed within 1 year (n=371) after Dx for comparison of genetic risk factors.
Results: The cohort included 15 females and 16 males, aged 21 -75 years (median: 60 years). Relapse occurred 3.0 -8.1 years after Dx (median: 4.0 yrs). According to MRC criteria, pts were assigned to the following cytogenetic risk groups (cytogenetic data was not available for 2 pts): good risk, n=4 (14%); intermediate risk, n=22 (76%) and adverse risk, n=3 (10%). FLT3 internal tandem duplication (ITD) was identified in 5 of 29 tested pts (17%). When comparing pts that relapse within the first year after Dx to our cohort of pts with relapse after >3 years, we observed a trend towards a lower frequency of high-risk genetic parameters in the latter group (adverse risk cytogenetics 18% vs 10%; FLT3-ITD, 32% vs 17%; TP53 mutation 8% vs 0%). However, this trend was not statistically significant. Cytogenetic changes between Dx and relapse were observed in 13/27 (48%) of pts analyzed at both time points. In 8/13 (61%) of these cases, a single cytogenetic aberration was gained at relapse, while a new complex karyotype was observed in 2/13 (15%) of relapsed pts and in 1 pt with complex karyotype at diagnosis, additional aberrations were detected at relapse. Furthermore, we identified cytogenetically independent clones between Dx and relapse in 2/13 (15%) cases.
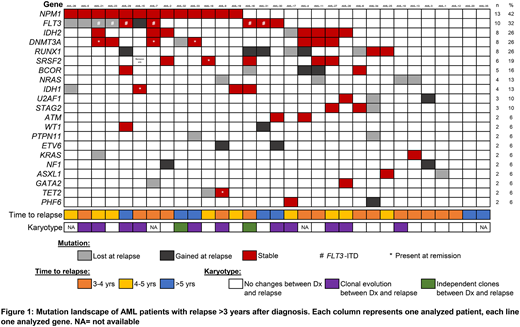
Next, we analyzed the molecular genetic evolution in our cohort. Overall, WES identified a total of 106 mutations in 30 genes associated with myeloid neoplasia (Figure 1). Following genes were mutated in >10% of pts: NPM1 (13/31 of pts), FLT3 (10/31), IDH2, DNMT3A and RUNX1 (8/31, each), SRSF2 (6/31), BCOR (5/31), NRAS and IDH1 (4/31, each). When comparing the matched diagnostic and relapse samples, a total of 61 mutations were detectable at both time points. A total of 24 mutations present at Dx were lost at relapse. Most frequently, mutations in NRAS (n=4) and FLT3 (n=4) were not detectable in the relapse samples. Overall, 12/24 (50%) of mutations lost at relapse affected signaling pathways, followed by transcription factors and epigenetic modifiers (n=5, each). In contrast, 21 mutations were gained during disease progression. Ten of these mutations (48%) are predicted to result in truncation of a transcription factor with known role in normal hematopoiesis (RUNX1, n=4; ETV6 and BCOR, n= 2; WT1 and BCORL1, n=1). In addition, 4 mutations gained during disease progression affected tumor suppressor genes (NF1, n=2; TP53, n=1, ATM, n=1). In 6/19 (31%) of pts with available data mutations in epigenetic modifiers persisted in remission (DNMT3A, n=3; IDH1, SRSF2, and TET2, n=1).
Conclusion: In our cohort of AML pts with relapses >3 years after Dx, we observed a high frequency of cases that lost mutations affecting signaling pathways during disease progression and gained mutations in hematopoietic transcription factors, indicating that the relapse clone in these patients is promoted by aberrant differentiation of hematopoietic stem cells.
Hartmann:MLL Munich Leukemia Laboratory: Employment. Nadarajah:MLL Munich Leukemia Laboratory: Employment. Meggendorfer:MLL Munich Leukemia Laboratory: Employment. Stengel:MLL Munich Leukemia Laboratory: Employment. Kern:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal