

Abstract
Introduction: Red blood cell (RBC) transfusions are central in the management of sickle cell disease (SCD), an inherited hemoglobinopathy characterized by hemolysis, acute pain, and multi-systemic complications. Extended matching of patient and donor RBC antigens is an established strategy to minimize alloimmunization, which can make provision of compatible blood difficult and can result in severe, even lethal hemolytic transfusion reactions. While RBC genotype matching has proven valuable in SCD transfusion practice, current technologies are often limited in throughput and focus on selected blood groups and known variants. Limited information is available comparing whole genome sequencing (WGS) with other blood typing platforms in SCD.
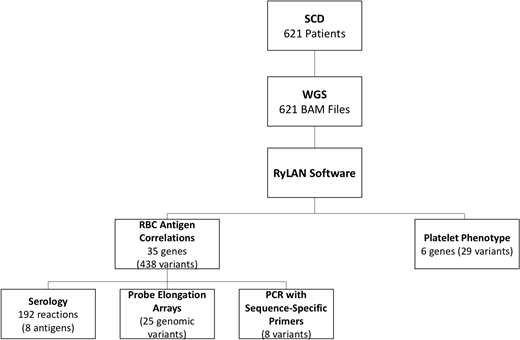
Design/Methods: WGS was performed on stored blood samples from 621 SCD patients recruited into two clinical studies. We utilized our open-source Python application (RyLAN), to translate WGS data into a predicted extended RBC and platelet phenotype. The 467 genomic variants interpreted by RyLAN in 41 genes were correlated with clinical and laboratory data in the immunohematology and electronic health records (Figure 1).
Results: The 621 patients included 485 HbSS, 21 HbSb0, 29 HbSβ+, 84 HbSC, 1HbS O Arab, and 1 HbSD. The mean age was 34.3 ± 12.1 years, and 54% were female. Health records indicated that 383 (62%) patients had previously received RBC transfusions and 17(3%) had never been transfused; the status of the remaining 221 was unknown.
RyLAN software was executed as a singularity container in multithreaded mode, completing the analysis of all 621 bam WGS files in 8.5 hours (8 CPUs and 16GB of memory per file). The average read depth for genomic positions of interest was 33 and the average QUAL value was 644. The highest variant allele frequency was detected at the Fyb, ACKR1 promoter, and the KCAM- loci (94%, 86% and 82%, respectively). Each of the 621 participants demonstrated a unique extended blood group genotype through WGS. RyLAN predicted 237 unique extended platelet phenotype combinations in this cohort, including HPA-25bw and HPA-13bw positive patients.
Blood antigen WGS predictions were correlated with other typing methods in 112 individuals: 192 total serologic reactions for 8 antigens; 55 documented alloantibodies; 25 genomic variants in 71 participants by probe-elongation array; and PCR with sequence-specific primers for 8 variants in 13 individuals (Figure 1). Two instances of heterozygosity (Jka/Jkb and Doa/Dob) were undetected due to low read depth, and 8 unresolved discrepancies were identified: 2 with serology, 1 with a reported historical alloantibody, 2 with probe-elongation array determinations, and 1 with the PCR method. WGS detected multiple weak blood group variants, surpassing the sensitivity of serology in one complex case, as well as rare phenotypes including 4 Yka-, 5 Kna/Knb, 1 FORS1-, and 2 Jra- cases. The algorithm correctly predicted an Sla-negative RBC phenotype in a patient with documented anti-Sla alloantibody.
Conclusion: We describe an efficient, open-source algorithm used to interpret 35 minor blood group and 6 platelet antigen genes from WGS in a large SCD cohort. Eight unresolved discrepancies were identified from 2126 correlation events with serology, alloimmunization history, and other genotyping methods in a subset of 112 individuals. WGS demonstrated higher sensitivity for weak antigen detection compared with serology, and a capacity to detect rare phenotypes not readily determined by other methods. Sanger resequencing is currently in progress to validate rare phenotype predictions and resolve remaining discrepancies. Future studies are needed to refine WGS algorithms in SCD, and determine the value of this technology for alloantibody identification, optimal blood group allocation, and donor recruitment.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal