

Abstract
Introduction: Oncogenic mutations of Ras genes have been implicated in number of human cancers whereby the cells grow uncontrollably, and evade death signals (Ledford, Nature, 520, 278, 2015). Rigosertib is a small molecule that inhibits cellular signaling in cancer cells by acting as a Ras mimetic (Athuluri-Divakar SK, Cell 2016). Rigosertib, as a single agent, was evaluated in a Phase III clinical trial in refractory MDS patients where the drug was administered as a 3 day continuous IV infusion every other week (NCT01241500). Rigosertib has also been evaluated in Phase I/II trials via oral route of administration in patients with solid malignancies (NCT01168011); and with myelodysplastic syndromes (NCT01926587). We performed pharmacokinetic and safety analysis of 253 patients across three different trials. The pharmacokinetics and safety profile of orally administered Rigosertib was evaluated in patients with MDS and solid malignancies. The results were compared to data obtained from high risk MDS patients receiving intravenous rigosertib therapy. In this investigation, we present evidence that differential GU toxicity of Rigosertib is probably related to the route of administration and dosing regimen; and independent of the underlying disease state.
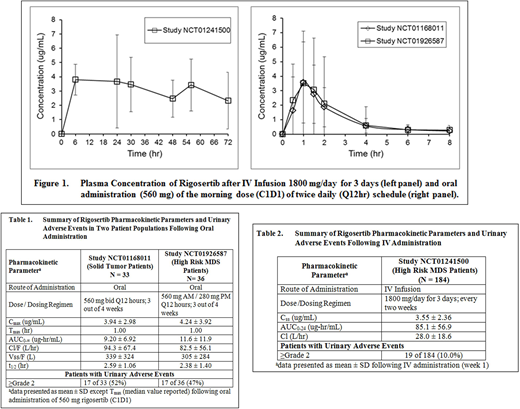
Methods: Data was analyzed from three clinical studies. Study NCT01241500 was conducted with MDS patients (n = 184) who were administered Rigosertib by continuous IV infusion (1800 mg/day for 72 hours) every 2 weeks. Blood samples were collected at 6, 24, 30, 48, 56 and 72 hours after infusion initiation. In Study NCT01168011, patients with advanced solid tumors (n=33) received oral Rigosertib (560 mg) Q12 hours of a 28-day cycle (3 weeks on, 1 week off). Study NCT01926587, conducted in MDS patients (n=36) with oral administration Rigosertib in combination with parenteral Azacitidine, evaluated the same AM dose but a lower 280 mg PM dose. In both studies, pharmacokinetic assessment was conducted after administration of the AM dose (560 mg) on day 1 of cycle 1. Blood samples were collected predose and 0.5, 1, 1.5, 2, 4, 6 and 8 hours after administration. For Study NCT01168011, urine was collected for 24 hours (0-4, 4-8 and 8-24 hrs) to determine the percent of dose excreted in urine. Rigosertib concentrations in plasma and urine were determined by a validated LC/MS-MS method. Rigosertib pharmacokinetic parameters were estimated using non-compartmental analysis. Treatment emergent adverse events were recorded throughout each study.
Results: There was no difference in Rigosertib pharmacokinetic parameters between the two patient populations (Table 1) upon oral administration, and the plasma concentration-time profiles were super imposable (Figure 1). Approximately 2% of the orally administered dose was recovered in the urine. In both studies, the most common AEs observed were GU AEs (dysuria, hematuria). The incidence of grade ≥2 GU toxicity observed with oral dosing was 17 of 33 (52%) and 17 of 36 (47%) in Study NCT01168011 and NCT01926587, respectively. The incidence of GU AEs in both the studies was comparable even though the PM dose was lower (560 mg vs 280mg) in study NCT01926587. Peak plasma concentration and AUC0-∞ were similar in patients with or without urinary symptoms. Compared to oral dosing, the incidences of GU AEs were dramatically lower (10%) following IV infusion despite 8-fold higher and more prolonged systemic exposure (Table 2).
Conclusions: This study demonstrates that the urinary adverse events and systemic exposure are similar in patients with either solid malignancies or MDS treated with orally administered rigosertib. The incidence of GU AEs is relatively high in spite of the very low amount (2%) of the administered dose eliminated through the renal route. In contrast, the GU AEs are significantly lower when Rigosertib is administered by IV infusion despite of 8 fold higher exposure. This is probably due to the short half-life of drug and dosing holiday of 12 days between two doses within the cycle. The higher incidence of GU AE upon oral administration is likely related to the dwell time of high concentration of drug in the bladder at night in patients treated daily for 3/4 weeks. This was successfully addressed in the clinical trial, NCT01926587 expansion with risk mitigation strategies, by deploying a novel dosing regimen derived through pharmacokinetic modeling and simulation (Taft, et al; ASH 2018 Abstract Submitted).
Maniar:Onconova Therapeutics, Inc: Employment, Equity Ownership. Ren:Onconova Therapeutics, Inc: Employment, Equity Ownership. Zbyszewski:Onconova Therapeutics, Inc: Employment, Equity Ownership. Petrone:Onconova Terapeutics Inc.: Employment, Equity Ownership. Fruchtman:Onconova Therapeutic Inc: Employment, Equity Ownership. Taft:Onconova Therapeutics, Inc: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal