

In this issue of Blood, 1 have identified that the N-terminal noncatalytic heavy chain of factor XII (FXII) plays an important role in the regulation of activation of the contact pathway of coagulation by the reciprocal activation of FXII and prekallikrein (PK). This mechanism provides insights into the clinical manifestation of an edema outbreak in patients with a rare form of hereditary angioedema (HAE).
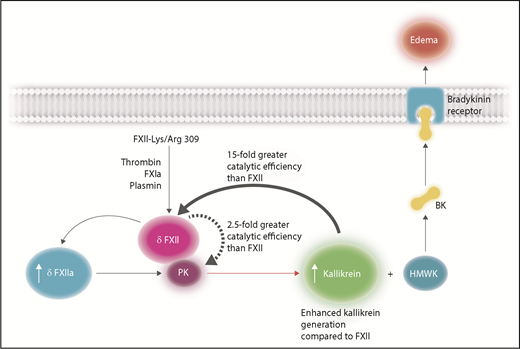
FXII-Lys/Arg309 results in enhanced kallikrein activation leading to bradykinin (BK) release from HMWK that is unable to be effectively inhibited by C1-INH. The variant FXII-Lys/Arg309 may be cleaved after residues 309 by thrombin, FXIa, and plasmin, in circumstances such as trauma, to give rise to a nonactivated truncated form of FXII, denoted δFXII, which contains the catalytic domain without the heavy chain of FXII. There are 2 mechanisms that give rise to enhanced kallikrein generation: first, δFXII can convert PK to kallikrein more efficiently than native FXII (with a 2.5-fold increase in catalytic efficiency) thereby generating more kallikrein in the early stages of reciprocal FXII/PK activation. Second, δFXII is a better substrate than FXII for kallikrein (15-fold greater catalytic efficiency). As a result, increased concentrations of δFXIIa will be generated, causing further PK activation, ultimately resulting in enhanced kallikrein generation. Kallikrein will cleave HMWK, releasing BK that will bind to BK receptors and induce vascular permeability leading to edema. Professional illustration by Somersault18:24.
FXII-Lys/Arg309 results in enhanced kallikrein activation leading to bradykinin (BK) release from HMWK that is unable to be effectively inhibited by C1-INH. The variant FXII-Lys/Arg309 may be cleaved after residues 309 by thrombin, FXIa, and plasmin, in circumstances such as trauma, to give rise to a nonactivated truncated form of FXII, denoted δFXII, which contains the catalytic domain without the heavy chain of FXII. There are 2 mechanisms that give rise to enhanced kallikrein generation: first, δFXII can convert PK to kallikrein more efficiently than native FXII (with a 2.5-fold increase in catalytic efficiency) thereby generating more kallikrein in the early stages of reciprocal FXII/PK activation. Second, δFXII is a better substrate than FXII for kallikrein (15-fold greater catalytic efficiency). As a result, increased concentrations of δFXIIa will be generated, causing further PK activation, ultimately resulting in enhanced kallikrein generation. Kallikrein will cleave HMWK, releasing BK that will bind to BK receptors and induce vascular permeability leading to edema. Professional illustration by Somersault18:24.
Most cases of HAE (type I and type II) arise by the inability of the naturally occurring inhibitor, C1-inhibitor (C1-INH) to inhibit activation of the contact pathway of coagulation, which results in high levels of kallikrein that can subsequently cleave high-molecular-weight kininogen (HMWK) to generate bradykinin. In turn, bradykinin binds to bradykinin receptors leading to increased vascular permeability and the presentation of edema. HAE may also arise in circumstances such as trauma in patients with normal levels of functionally active C1-INH (type III); one such example is caused by a variant of FXII whereby Thr309 is substituted with a lysine or arginine (FXII-Lys/Arg309).2 A question that remains to be answered is why this variant results in edema when C1-INH is present at normal levels and fully functional. Björkqvist et al3 have characterized that the FXII Lys/Arg309 variants exhibit defective glycosylation, and this leads to a FXII that is more readily activated in the presence of a surface. Ivanov et al have further identified that the presence of the heavy chain plays a role in the regulation of solution-phase activation of PK and FXII. By preparing recombinant FXII variants, the authors have characterized that FXII-Lys/Arg309 is cleaved after residue 309 by both thrombin and activated factor XI (FXIa) to produce a truncated form of FXII that remains a zymogen and no longer has the noncatalytic heavy chain attached (designated δFXII). There are then 2 mechanisms (see figure) by which δFXII can subsequently enhance the activation of the kallikrein-kinin pathway. The first is that the δFXII can convert PK to kallikrein more efficiently than native FXII (with a 2.5-fold increase in catalytic efficiency), thereby generating more kallikrein in the early stages of reciprocal FXII/PK activation. The second mechanism is that δFXII is a better substrate than FXII for kallikrein (15-fold greater catalytic efficiency). The accelerated FXII/PK activation is too much for normal active C1-INH to inhibit immediately, and as a consequence, the free kallikrein will cleave HMWK to generate bradykinin with consequential edema in patients, with the FXII-Lys/Arg309 variant.
FXII comprises an N-terminal noncatalytic domain (known as heavy chain) and a catalytic domain (also known as the light chain). Regions of the heavy chain have been implicated in many binding interactions.4 The plasma contact pathway is initiated by reactions between FXII, PK, and HMWK, leading to activated FXII (FXIIa) and kallikrein. Also, upon binding to negatively charged surfaces, FXII can undergo autoactivation.5 Recently, Ivanov et al have demonstrated that zymogen FXII has “zymogen activity” that is sufficient to initiate reciprocal activation of PK and FXII,6 a mechanism that is enhanced by a surface. There are 3 potential cleavage sites on FXII, located at Arg334, Arg343, and Arg353. Cleavage of FXII at Arg353 is necessary to yield 2-chain active FXIIa (αFXIIa).7 Subsequent cleavage at Arg334 releases the heavy chain, leaving the truncated active catalytic domain of FXII, βFXIIa, which no longer possesses the ability to bind to prothrombotic surfaces and is therefore considered to be more selective toward the activation of PK.8
Recently, de Maat et al9 identified that truncation of FXII primes it for activation by kallikrein in solution and proposed that the heavy chain of FXII shields its activating cleavage site, Arg353. Therefore, naturally occurring truncation of FXII (that may occur in HAE FXII-Lys/Arg309 via plasmin cleavage10 ) exposes the Arg353, making it more susceptible to activation. Within this issue of Blood, Ivanov et al have extended these studies and demonstrated that the FXII-Lys/Arg309 variant can also be cleaved by FXIa and thrombin at the same position as plasmin. These cleavages give rise to a truncated form of the catalytic domain (termed δFXII) that does not possess any catalytic activity alone, yet in the presence of PK shows enhanced activation of PK compared with full-length FXII zymogen to give enhanced kallikrein generation. Furthermore, δFXII is a better substrate than FXII for kallikrein, exhibiting a 15-fold greater catalytic efficiency for generating δFXIIa compared with the activation of FXII. As proposed by de Maat et al,9 truncated FXII is more likely to expose Arg353, making it more susceptible to cleavage by kallikrein in a non-surface–dependent manner. It is likely that binding of FXII to surfaces modifies the conformation of FXII to make Arg353 more exposed for cleavage. Interestingly, Ivanov et al found that the antibody 15H8 that binds to the heavy chain of FXII produces the same effects as δFXII, giving rise to enhanced reciprocal activation of PK/FXII when bound to full-length wild-type FXII.
The current study by Ivanov et al provides further explanation as to why patients with FXII-Lys/Arg309 may exhibit trauma-induced edema. Essentially, trauma will give rise to elevated levels of FXIa, thrombin, and plasmin, all of which are capable of cleaving FXII after residues Lys/Arg309. As a consequence, there will be an enhancement in the activation of the contact pathway by increased reciprocal δFXII/PK zymogen activation and more efficient generation of δFXIIa by kallikrein. Ultimately, the consequence is rapid generation of kallikrein that is not able to be effectively inhibited by C1-INH and therefore gives rise to bradykinin formation. This study raises the question of whether fast-acting small-molecule or antibody inhibitors of kallikrein or FXIIa may result in improved therapy for edema in patients with the FXII-Lys/Arg309 variant. Indeed, an active site inhibitor of FXIIa exhibits very encouraging preclinical data for the potential treatment of type III HAE.3
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal