

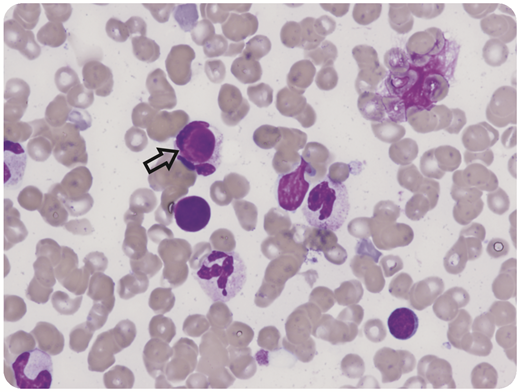
An 8-year-old girl presented with fever, lymphadenopathy, oral ulcers, and gum oozing. She was found to have pancytopenia and prolonged prothrombin time/activated partial thromboplastin time suspicious for acute promyelocytic leukemia. Bone marrow examination showed no evidence of malignancy; however, lupus erythematosus (LE) cells (macrophages that have phagocytized the denatured nucleus of another cell) were noted in the aspirate (black arrow, original magnification ×60, hematoxylin and eosin stain). The patient also tested positive for antiphospholipid antibodies: anticardiolipin immunoglobulin G 75.8 GPL (<23 GPL) and anti-β-2 glycoprotein immunoglobulin G 63.1 GPL (<20.1 GPL). Dilute Russell viper venom time ratio was 1.79 (<1.19), and Staclot difference was 78.8 seconds (<10.7 seconds). Prothrombin level was 12% (reference range 50%-150%), with positive antiprothrombin immunoglobulin G 140.9 units (0-19.9 units). The patient was diagnosed with systemic LE (11/17 Systemic Lupus International Collaborating Clinics criteria) and lupus anticoagulant hypoprothrombinemia syndrome (LAHPS). LE cells are characteristic of autoimmune conditions, although they are not specific to LAHPS or systemic LE.
LAHPS is characterized by the presence of a lupus anticoagulant and an antibody to prothrombin, resulting in both hemorrhagic and thrombotic symptoms. In general, patients respond well to prothrombin complex concentrate, corticosteroids, rituximab, and/or intravenous immunoglobulin. Despite these interventions and a prothrombin level above 50%, the patient died from a large intracranial hemorrhage. The case illustrates a helpful microscopic finding in the bone marrow that may suggest the diagnosis of LAHPS.
An 8-year-old girl presented with fever, lymphadenopathy, oral ulcers, and gum oozing. She was found to have pancytopenia and prolonged prothrombin time/activated partial thromboplastin time suspicious for acute promyelocytic leukemia. Bone marrow examination showed no evidence of malignancy; however, lupus erythematosus (LE) cells (macrophages that have phagocytized the denatured nucleus of another cell) were noted in the aspirate (black arrow, original magnification ×60, hematoxylin and eosin stain). The patient also tested positive for antiphospholipid antibodies: anticardiolipin immunoglobulin G 75.8 GPL (<23 GPL) and anti-β-2 glycoprotein immunoglobulin G 63.1 GPL (<20.1 GPL). Dilute Russell viper venom time ratio was 1.79 (<1.19), and Staclot difference was 78.8 seconds (<10.7 seconds). Prothrombin level was 12% (reference range 50%-150%), with positive antiprothrombin immunoglobulin G 140.9 units (0-19.9 units). The patient was diagnosed with systemic LE (11/17 Systemic Lupus International Collaborating Clinics criteria) and lupus anticoagulant hypoprothrombinemia syndrome (LAHPS). LE cells are characteristic of autoimmune conditions, although they are not specific to LAHPS or systemic LE.
LAHPS is characterized by the presence of a lupus anticoagulant and an antibody to prothrombin, resulting in both hemorrhagic and thrombotic symptoms. In general, patients respond well to prothrombin complex concentrate, corticosteroids, rituximab, and/or intravenous immunoglobulin. Despite these interventions and a prothrombin level above 50%, the patient died from a large intracranial hemorrhage. The case illustrates a helpful microscopic finding in the bone marrow that may suggest the diagnosis of LAHPS.
For additional images, visit the ASH Image Bank, a reference and teaching tool that is continually updated with new atlas and case study images. For more information, visit http://imagebank.hematology.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal