

In this issue of Blood, report a phase 1/2 study of voxelotor (GBT440) in sickle cell anemia. This drug binds to the α-globin chain of hemoglobin and increases hemoglobin-oxygen affinity, thereby decreasing the polymerization tendency of deoxygenated sickle hemoglobin (HbS). Treated patients had a median 1 g/dL increase of hemoglobin concentration, and markers of hemolysis were decreased.1
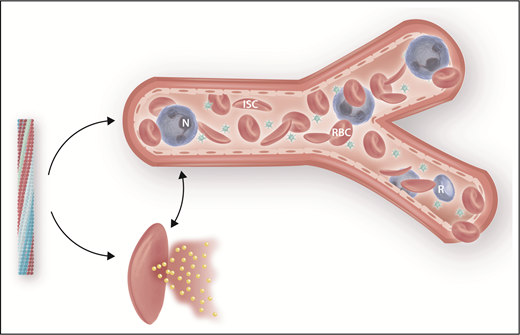
Deoxygenated HbS polymerization is the primary trigger of sickle cell disease pathophysiology, and its inhibition is therapeutically important. Polymerization (left) can be averted by (1) increasing HbF levels; (2) increasing hemoglobin-oxygen affinity, as with voxelator; (3) “rehydrating” the sickle cell, thereby decreasing the concentration of HbS.9 Polymer-damaged sickle erythrocytes cause vaso-occlusion (top) and hemolysis, a portion of which occurs intravascularly (bottom). ISC, irreversibly sickle cell; N, neutrophil; R, reticulocyte; RBC, sickle erythrocyte. Professional illustration by Somersault18:24.
Deoxygenated HbS polymerization is the primary trigger of sickle cell disease pathophysiology, and its inhibition is therapeutically important. Polymerization (left) can be averted by (1) increasing HbF levels; (2) increasing hemoglobin-oxygen affinity, as with voxelator; (3) “rehydrating” the sickle cell, thereby decreasing the concentration of HbS.9 Polymer-damaged sickle erythrocytes cause vaso-occlusion (top) and hemolysis, a portion of which occurs intravascularly (bottom). ISC, irreversibly sickle cell; N, neutrophil; R, reticulocyte; RBC, sickle erythrocyte. Professional illustration by Somersault18:24.
Hydroxyurea is the only approved drug that modifies the course of sickle cell disease. Based on controlled clinical trials in children and adults, it has become the standard of care for this disease, and nearly all patients should receive this drug starting in the first 2 years of life.2 It works primarily by inducing higher levels of fetal hemoglobin (HbF). HbF prevents HbS polymerization because of its exclusion from the polymer. Although most patients, especially when they are young, have an increase in HbF, the benefits of hydroxyurea are not uniform, and 5% to 15% of patients, mainly adults, do not respond or respond inadequately. Voxelator, because it also directly effects HbS polymerization, is another potential disease-modifying treatment. If the results reported here are confirmed, this agent could be employed with hydroxyurea in a combination-therapy approach targeting HbS polymerization; it might even be used as stand-alone treatment in patients who do not respond to or cannot take hydroxyurea.
Sickle cell disease pathophysiology is triggered by HbS polymerization (see figure). Erythrocytes damaged by HbS polymer lead directly and indirectly to sickle vaso-occlusion and hemolytic anemia.3 Many clinical subphenotypes characterize sickle cell disease, and although vaso-occlusion and hemolysis are interrelated at some level, the complications of disease tend to be associated with 1 or the other of these 2 pathophysiologic branches. Hemolytic anemia, particularly that component of hemolysis that occurs intravascularly, is associated with a suite of complications that develop over years and include pulmonary and systemic vascular disease, cerebrovascular disease, and sickle nephropathy, and patients with the most intense hemolysis have increased mortality.4 Reducing the rate of hemolysis, as appears to be the case with voxelator, is consequently an attractive approach to treatment.
However, there is reason for caution. Higher hemoglobin levels might have adverse effects on the complications of disease closely linked to sickle vaso-occlusion. Perhaps this is a result of increased blood viscosity due to the longer survival of erythrocytes that still contain mainly HbS. Compared with simple homozygotes for the HbS gene, the one-third of sickle cell anemia patients who have α thalassemia in addition to sickle cell anemia have ∼0.5 to 1 g/dL higher hemoglobin levels because they have less hemolysis due to reduced mean cell HbS concentration. Nevertheless, they have an increased risk of certain vaso-occlusive viscosity-related complications like osteonecrosis and painful episodes. Beneficially, they have a reduced propensity to stroke, sickle nephropathy, leg ulcers, and priapism.5 These observations were reinforced by the results of a phase 3 clinical trial of senicapoc, an inhibitor of potassium traffic across the erythrocyte membrane via the Gardos channel. The “rehydrated” sickle erythrocytes of treated patients had reduced HbS concentration, and like patients with sickle cell anemia–α thalassemia, hemolysis was decreased and hemoglobin concentration increased by ∼0.6 g/dL. Unfortunately, the trial had to be stopped prematurely as an interim analysis portended futility.6 In some subgroups, drug-treated patients had an increased rate of vaso-occlusive events. The short duration of this trial made it unlikely that any improvement of the hemolysis-associated complications of disease would be observed. Similarly, it might take years before any benefit of voxelator on these complications can be appreciated. In contrast to the observations in sickle cell anemia–α thalassemia, although hemolysis is reduced and hemoglobin is increased by 0.5 to 1 g/dL when patients are treated with hydroxyurea, the rate of acute sickle vaso-occlusive events falls. This could be the result of direct inhibition of HbS polymerization by HbF compared with the indirect effects of improved cell hydration where there is no hemoglobin modification. It remains unclear how the increased hemoglobin level that results from voxcelator treatment will affect sickle vaso-occlusive events. The optimistic view is that as this agent directly modifies hemoglobin as does HbF, higher hemoglobin levels that result from treatment will not favor vaso-occlusion.
This study included 40 patients who received active drug for 4 to 12 weeks. Only 4 continued treatment of up to 6 months, so long-term safety cannot be assumed. Regulation of oxygen delivery is a fundamental physiologic process, and peril could lurk if this is compromised. Although exercise testing results did not differ between treated cases and controls and oxygen delivery seemed unimpaired, longer-term adverse effect of increased hemoglobin-oxygen affinity must be considered. This is especially important when this drug is given to young children with developing nervous systems that are highly dependent on optimal oxygen delivery. In sickle cell anemia, blood flow to the brain is already maximal, and the presence of hemoglobin unable to unload oxygen could have an adverse outcome.7 Some rare hemoglobin variants have intrinsically increased oxygen affinity with p50 levels less than those caused by voxcelator. Clinically, these patients seem fine, but they do not have the damaged red blood cells and vasculopathy of sickle cell anemia. HbF has higher oxygen affinity than HbS, but patients with drug-induced or naturally high HbF levels do not appear to be adversely affected. Compound heterozygotes for HbS and gene deletion hereditary persistence of HbF are well despite having ∼30% HbF in all of their erythrocytes.8
Sadly, drugs that impact the progression of sickle cell anemia are limited to hydroxyurea that was approved >2 decades ago. If the preliminary results of voxcelator treatment are replicated in phase 3 clinical trials, the availability of an additional agent directly targeting polymerization, which reduces hemolysis and perhaps the long-term vasculopathic complications of hemolysis, would be major therapeutic progress.9
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal