

Abstract
Transfusion-associated circulatory overload (TACO) and transfusion-related acute lung injury (TRALI) are syndromes of acute respiratory distress that occur within 6 hours of blood transfusion. TACO and TRALI are the leading causes of transfusion-related fatalities, and specific therapies are unavailable. Diagnostically, it remains very challenging to distinguish TACO and TRALI from underlying causes of lung injury and/or fluid overload as well as from each other. TACO is characterized by pulmonary hydrostatic (cardiogenic) edema, whereas TRALI presents as pulmonary permeability edema (noncardiogenic). The pathophysiology of both syndromes is complex and incompletely understood. A 2-hit model is generally assumed to underlie TACO and TRALI disease pathology, where the first hit represents the clinical condition of the patient and the second hit is conveyed by the transfusion product. In TACO, cardiac or renal impairment and positive fluid balance appear first hits, whereas suboptimal fluid management or other components in the transfused product may enable the second hit. Remarkably, other factors beyond volume play a role in TACO. In TRALI, the first hit can, for example, be represented by inflammation, whereas the second hit is assumed to be caused by antileukocyte antibodies or biological response modifiers (eg, lipids). In this review, we provide an up-to-date overview of TACO and TRALI regarding clinical definitions, diagnostic strategies, pathophysiological mechanisms, and potential therapies. More research is required to better understand TACO and TRALI pathophysiology, and more biomarker studies are warranted. Collectively, this may result in improved diagnostics and development of therapeutic approaches for these life-threatening transfusion reactions.
Historical and current perspective
The occurrence of transfusion-associated circulatory overload (TACO) first received attention in the 1930s. In 1936, Plummer reported 5 fatalities due to circulatory overload after blood transfusion1 followed by more publications by Pygott2 and DeGowin3 in the 1930s, and Drummond4 and Pelner and Waldman5 in the 1940s and 1950s. Pulmonary edema was observed in these cases, and patients with left-sided heart disease were found to be at particular risk.4,5 Despite this early awareness, it was not until the 1990s when TACO received renewed attention and was seen as a distinct clinical entity.6 Currently, TACO is the most frequent pulmonary complication of transfusion, and it is an independent risk factor for in-hospital morbidity and mortality with increased incidence in a mixed intensive care unit population.7 The estimated frequency of TACO varies from 1% in hemovigilance reports, up to 8% in postoperative elderly patients, and up to 11% in critically ill patients.8-10 Using a prospective active surveillance algorithm, the incidence of TACO was found to be 1%.11 In a pediatric intensive care population consisting of 136 patients, the incidence of TACO was estimated to be between 1.5% and 11%.12 The National Blood Collection and Utilization Survey report described TACO to occur in 1:14 000 transfused components.13 TACO appears to be one of the leading causes of transfusion-related fatalities, with 44.1% of the reported transfusion-related fatalities being from TACO (60/136 reported transfusion-related deaths from 2010 to 2017) according to the Serious Hazards of Transfusion (SHOT) report.14 The Food and Drug Administration (FDA) reported that 30% of the reported transfusion-related fatalities were due to TACO (56/186 reported transfusion-related deaths from 2012 to 2016).15
Transfusion-related acute lung injury (TRALI) was first reported in 1951 by Barnard, who described a patient with acute leukemia who died upon blood transfusion due to an acute pulmonary reaction due to a hypersensitivity response.16 In 1957, Brittingham also described a TRALI case of a severe pulmonary reaction due to transfused blood containing high-titer leukoagglutinins.17 In 1966, Phillips et al reported 3 transfused patients who suffered from pulmonary edema without fluid overfload.18 In 1970, Ward observed the occurrence of noncardiogenic pulmonary edema possibly due to antileukocyte antibodies,19 and in 1971, Thompson et al published a case report concerning a fatal pulmonary reaction to HLA-incompatible blood transfusion.20 It was not until 1983 when TRALI was officially recognized as a separate disease entity because it was described to occur due to passive transfer of antileukocyte antibodies.21 The National Blood Collection and Utilization Survey reported TRALI to occur 1:64 000 transfused components,13 and the frequency of TRALI has been estimated to be ∼0.08% to 15.1% per patient and 0.01% to 1.12% per product.22 The FDA has reported TRALI to be the leading cause of transfusion-related fatalities for many years with 34% of the reported transfusion-related fatalities being from TRALI (64/186 reported transfusion-related deaths from 2012 to 2016).15 In contrast, the 2017 SHOT report published that ∼4% of the reported transfusion-related fatalities were due to TRALI (5/136 reported transfusion-related deaths from 2010 to 2017).14 Survival of TRALI in critically ill patients has been reported to be as low as 53% compared with 83% in acute lung injury control patients.23
Clinical presentation and diagnosis
TACO and TRALI are life-threatening transfusion reactions, and it remains challenging to accurately diagnose and distinguish both syndromes. Both TACO and TRALI present with the onset of acute respiratory distress (hypoxemia) within 6 hours of a blood transfusion and demonstrate infiltrates on a frontal chest radiograph indicative of the presence of pulmonary edema (Figure 1). Clinical definitions have been established for both syndromes (Table 1). For TACO, this includes the National Healthcare Safety Network definitions published in 201624 or the 2011 International Society of Blood Transfusion (ISBT) criteria,25 whereas for the diagnosis of TRALI, the 2005 National Heart, Lung, and Blood Institute Working Group definitions26 or the 2004 Canadian Consensus Conference criteria27 are commonly used (Table 1). The clinical definitions of TACO may include evidence of positive fluid balance or cardiogenic involvement, which may manifest as left heart failure, elevated blood pressure, or tachycardia. In contrast, TRALI is strictly noncardiogenic, without evidence of left arterial hypertension, and thus, circulatory overload must be excluded. In addition, no temporal relationship to an alternative risk factor for acute lung injury (eg, pneumonia, sepsis, aspiration, multiple trauma, and acute pancreatitis) may be present for the diagnosis of TRALI; otherwise, it has to be classified as “possible TRALI.” It has been suggested that the transfusion itself may have a minor contribution in possible TRALI and that this term should therefore be replaced by “transfused acute respiratory distress syndrome (ARDS).”28,29 Others, however, argue against this by stating that Possible TRALI is a distinct clinical entity, which is especially evident in critically ill and injured patients.30 Despite the similarity in clinical presentation between TACO and TRALI, key diagnostic features have been identified that may aid in clinical diagnosis and differentiation of TACO vs TRALI (Table 2).31 Unfortunately, the tools to perform these diagnostic analyses may not be widely available. These key diagnostic features include assessment of hydrostatic pulmonary pressure (increased in TACO), protein levels in edema fluid (protein poor in TACO), response to diuretics (may occur in TACO), and cardiogenic nonlaboratory parameters, which may be impaired in TACO (eg, decreased systolic injection fraction, increased systolic blood pressure, increased vascular pedicle width, and increased cardiothoracic ratio on chest radiograph, in TACO). Increased ventricular filling or myocardial stretching may be assessed as well in the diagnosis of TACO by analyzing the levels of BNP or N-terminal pro-BNP because both have demonstrated a significant positive correlation between pre- and posttransfusion levels in TACO patients.32,33 Caution, however, is required with the use of natriuretic peptides as a diagnostic measure for distinguishing TACO from TRALI, because these levels may also be significantly increased in critically ill patients suffering from TRALI.34 Importantly, cardiac ischemia (ischemic changes on electrocardiography or increased new troponin T levels) must be excluded in diagnosing TACO. Supportive clinical and laboratory features may perhaps further help in the diagnosis of TACO or TRALI (Table 3).35 This includes physical examination of neck veins (distended in TACO), auscultation (rales, S3 in TACO), and blood pressure measurement (increased in TACO). Regarding body temperature, TRALI patients are frequently febrile, but fever may be present in one-third of TACO patients as well.36 Supportive laboratory investigations may include determination of white blood cell counts (transient leukopenia and mild thrombocytopenia in TRALI),37-40 the detection of antibodies against recipient HLA class I/II and/or human neutrophil antigen (HNA) antigens in the involved blood donor (in ∼80% of TRALI cases),41 and analysis of cytokines, as is discussed later in more detail. It remains, however, unknown if blood cell counts are affected and if antileukocyte antibodies could be present in TACO patients. Notably, there are ongoing international efforts to further define TACO, which include the 2017 ISBT working party on Haemovigilance together with the International Haemovigilance Network, which have proposed revised TACO criteria to establish a surveillance definition for reporting and tracking purposes.42

Chest radiographs of a TACO and TRALI patient. Patient at time of TACO occurrence (A), and the same patient after resolution of TACO (B). Pretransfusion patient (C), and the same patient at time of TRALI occurrence (D). Normal chest radiographs without signs of pulmonary edema (B-C); infiltrative changes indicative of pulmonary edema (A,D). Adapted from Agnihotri and Agnihotri110 and Vlaar and Juffermans22 with permission.
Chest radiographs of a TACO and TRALI patient. Patient at time of TACO occurrence (A), and the same patient after resolution of TACO (B). Pretransfusion patient (C), and the same patient at time of TRALI occurrence (D). Normal chest radiographs without signs of pulmonary edema (B-C); infiltrative changes indicative of pulmonary edema (A,D). Adapted from Agnihotri and Agnihotri110 and Vlaar and Juffermans22 with permission.
Clinical definitions for TACO and TRALI
| NHSN 2016-TACO definition24 | ISBT 2011-TACO definition25 | NHLBI Working Group 2005-TRALI definition26 | Canadian Consensus Conference 2004-TRALI definition27 |
|---|---|---|---|
| New onset or exacerbation of ≥3 of the following within 6 h of transfusion: | Any 4 of the following occurring within 6 h of completion of transfusion: | Patients without acute lung injury (ALI) risk factor(s) other than transfusion: | ALI: |
| (a) Acute respiratory distress (dyspnea, orthopnea, and cough) | (a) Acute respiratory distress | In patients with no ALI immediately before transfusion, a temporal association of transfusion and ALI is made if there is: | (a) Acute onset |
| (b) Evidence of positive fluid balance | (b) Tachycardia | (a) New ALI,96 and | (b) Hypoxemia: PaO2/FiO2 ≤ 300, or SpO2 < 90% on room air (or other clinical evidence of hypoxemia in a nonresearch setting) |
| (c) Elevated brain natriuretic peptide | (c) Elevated blood pressure | (b) The onset of symptoms or signs is during or within 6 h after the end of transfusion of 1 or more plasma-containing blood products | (c) Bilateral infiltrates on frontal chest radiograph |
| (d) Radiographic evidence pulmonary edema | (d) Acute or worsening pulmonary edema of frontal chest radiograph | (d) No evidence of left atrial hypertension (ie, circulatory overload) | |
| (e) Evidence of left heart failure | (e) Evidence of positive fluid balance | As there is no other ALI risk factor, the new ALI is inferred to be mechanistically related to transfusion (ie, TRALI) | No preexisting ALI before transfusion |
| (f) Elevated central venous pressure | Patients with ALI risk factor(s) other than transfusion: | During or within 6 h of transfusion | |
| In patients with no ALI immediately before transfusion, a temporal association of transfusion and ALI is made if there is: | No temporal relationship to an alternative risk factor for ALI | ||
| (a) New ALI,96 and | If clear temporal relationship to an alternative risk factor for ALI (in combination with previous listed criteria), then defined as “possible TRALI” | ||
| (b) The onset of symptoms or signs is during or within 6 h after the end of transfusion of 1 or more plasma-containing blood products | |||
| By assessing the patient’s clinical course, the new ALI is either: | |||
| (a) TRALI, and the new ALI is inferred to be mechanistically related to the transfusion, or both the transfusion and the alternative risk factor, or | |||
| (b) Not TRALI, and the new ALI is mechanistically related to the alternative ALI risk factor alone, whereas the transfusion is coincidental |
| NHSN 2016-TACO definition24 | ISBT 2011-TACO definition25 | NHLBI Working Group 2005-TRALI definition26 | Canadian Consensus Conference 2004-TRALI definition27 |
|---|---|---|---|
| New onset or exacerbation of ≥3 of the following within 6 h of transfusion: | Any 4 of the following occurring within 6 h of completion of transfusion: | Patients without acute lung injury (ALI) risk factor(s) other than transfusion: | ALI: |
| (a) Acute respiratory distress (dyspnea, orthopnea, and cough) | (a) Acute respiratory distress | In patients with no ALI immediately before transfusion, a temporal association of transfusion and ALI is made if there is: | (a) Acute onset |
| (b) Evidence of positive fluid balance | (b) Tachycardia | (a) New ALI,96 and | (b) Hypoxemia: PaO2/FiO2 ≤ 300, or SpO2 < 90% on room air (or other clinical evidence of hypoxemia in a nonresearch setting) |
| (c) Elevated brain natriuretic peptide | (c) Elevated blood pressure | (b) The onset of symptoms or signs is during or within 6 h after the end of transfusion of 1 or more plasma-containing blood products | (c) Bilateral infiltrates on frontal chest radiograph |
| (d) Radiographic evidence pulmonary edema | (d) Acute or worsening pulmonary edema of frontal chest radiograph | (d) No evidence of left atrial hypertension (ie, circulatory overload) | |
| (e) Evidence of left heart failure | (e) Evidence of positive fluid balance | As there is no other ALI risk factor, the new ALI is inferred to be mechanistically related to transfusion (ie, TRALI) | No preexisting ALI before transfusion |
| (f) Elevated central venous pressure | Patients with ALI risk factor(s) other than transfusion: | During or within 6 h of transfusion | |
| In patients with no ALI immediately before transfusion, a temporal association of transfusion and ALI is made if there is: | No temporal relationship to an alternative risk factor for ALI | ||
| (a) New ALI,96 and | If clear temporal relationship to an alternative risk factor for ALI (in combination with previous listed criteria), then defined as “possible TRALI” | ||
| (b) The onset of symptoms or signs is during or within 6 h after the end of transfusion of 1 or more plasma-containing blood products | |||
| By assessing the patient’s clinical course, the new ALI is either: | |||
| (a) TRALI, and the new ALI is inferred to be mechanistically related to the transfusion, or both the transfusion and the alternative risk factor, or | |||
| (b) Not TRALI, and the new ALI is mechanistically related to the alternative ALI risk factor alone, whereas the transfusion is coincidental |
NHLBI, National Heart, Lung and Blood Institute; NHSN, National Healthcare Safety Network.
Key diagnostic features differentiating TACO from TRALI
| Key diagnostic feature | Specific diagnostic readout | TACO | TRALI |
|---|---|---|---|
| Acute onset of respiratory distress symptoms | Onset <6 h upon blood transfusion | Yes | Yes |
| Hypoxemia | SpO2 < 90% or PaO2/FiO2 < 300 mm Hg on room air | Yes | Yes |
| Pulmonary edema | Bilateral infiltrates on chest radiograph | Yes | Yes |
| Alternative risk factors for ALI | eg, pneumonia, sepsis, aspiration, multiple trauma, acute pancreatitis | No | No |
| Yes: possible TRALI | |||
| Hydrostatic pulmonary pressure increased | Pulmonary artery occlusion pressure >18 mm Hg | Yes | No |
| Protein-poor edema fluid | Edema or plasma protein concentration <0.65 at the onset of acute respiratory failure | Yes | No |
| Increased ventricular filling/myocardial stretching32-34 | B-type natriuretic peptide (BNP) >250 or pre-/posttransfusion BNP ratio >1.5 or N-terminal pro-BNP >1000 pg/mL | Yes | Yes*/No |
| Response to diuretics | Rapid and significant improvement | Yes | No |
| Cardiogenic nonlaboratory evidence for circulatory overload | Systolic ejection fraction <45 and no severe valvular heart disease on echocardiography | Yes | No |
| Systolic blood pressure >160 | |||
| Vascular pedicle width >65 mm and cardiothoracic ratio >0.55 on chest radiograph | |||
| Cardiac ischemia | New ischemic changes on electrocardiography or new troponin T levels of >0.05 | No | No |
| Key diagnostic feature | Specific diagnostic readout | TACO | TRALI |
|---|---|---|---|
| Acute onset of respiratory distress symptoms | Onset <6 h upon blood transfusion | Yes | Yes |
| Hypoxemia | SpO2 < 90% or PaO2/FiO2 < 300 mm Hg on room air | Yes | Yes |
| Pulmonary edema | Bilateral infiltrates on chest radiograph | Yes | Yes |
| Alternative risk factors for ALI | eg, pneumonia, sepsis, aspiration, multiple trauma, acute pancreatitis | No | No |
| Yes: possible TRALI | |||
| Hydrostatic pulmonary pressure increased | Pulmonary artery occlusion pressure >18 mm Hg | Yes | No |
| Protein-poor edema fluid | Edema or plasma protein concentration <0.65 at the onset of acute respiratory failure | Yes | No |
| Increased ventricular filling/myocardial stretching32-34 | B-type natriuretic peptide (BNP) >250 or pre-/posttransfusion BNP ratio >1.5 or N-terminal pro-BNP >1000 pg/mL | Yes | Yes*/No |
| Response to diuretics | Rapid and significant improvement | Yes | No |
| Cardiogenic nonlaboratory evidence for circulatory overload | Systolic ejection fraction <45 and no severe valvular heart disease on echocardiography | Yes | No |
| Systolic blood pressure >160 | |||
| Vascular pedicle width >65 mm and cardiothoracic ratio >0.55 on chest radiograph | |||
| Cardiac ischemia | New ischemic changes on electrocardiography or new troponin T levels of >0.05 | No | No |
Modified from Gajic et al.31
In transfused critically ill patients.
Supportive clinical and laboratory features further differentiating TACO from TRALI
| Supportive clinical and laboratory features | TACO | TRALI |
|---|---|---|
| Neck veins | Distended | Normal |
| Auscultation | Rales, S3 | Rales, No S3 |
| Blood pressure | Hypertension | Hypotension |
| Body temperature | Fever may be present (1/3 of patients)36 | Febrile |
| White blood cell count | Unknown | Transient leukopenia (infrequent) and mild thrombocytopenia37-40 |
| Leukocyte antibodies | Unknown | May be present (±80%)41 : |
| Anti-HLA class I antibodies | ||
| Anti-HLA class II antibodies | ||
| Anti-HNA antibodies | ||
| Posttransfusion cytokines49-51,59 | IL-6 increased | IL-6 increased |
| IL-8 not increased | IL-8 increased | |
| IL-10 increased | IL-10 not increased | |
| No changes in TNF-α, GM-CSF | No changes in TNF-α, GM-CSF |
| Supportive clinical and laboratory features | TACO | TRALI |
|---|---|---|
| Neck veins | Distended | Normal |
| Auscultation | Rales, S3 | Rales, No S3 |
| Blood pressure | Hypertension | Hypotension |
| Body temperature | Fever may be present (1/3 of patients)36 | Febrile |
| White blood cell count | Unknown | Transient leukopenia (infrequent) and mild thrombocytopenia37-40 |
| Leukocyte antibodies | Unknown | May be present (±80%)41 : |
| Anti-HLA class I antibodies | ||
| Anti-HLA class II antibodies | ||
| Anti-HNA antibodies | ||
| Posttransfusion cytokines49-51,59 | IL-6 increased | IL-6 increased |
| IL-8 not increased | IL-8 increased | |
| IL-10 increased | IL-10 not increased | |
| No changes in TNF-α, GM-CSF | No changes in TNF-α, GM-CSF |
Modified from Skeate et al.35
Pathophysiology
Two-hit model
The pathogeneses of TACO and TRALI are incompletely understood. As a general pathophysiological framework, a 2-hit model has been suggested for both TACO7 and TRALI.43,44 The first hit represents the underlying, preexisting clinical condition of the patient (transfusion-recipient risk factors), whereas the second hit is conveyed by the transfused blood product. Both hits are required for development of TACO or TRALI. The first hit in TACO may be represented by the poor adaptability for volume overload. A recent large prospective study enrolling 200 patients with TACO identified congestive heart failure, cardiomegaly on chest radiograph, pretransfusion diuretic use, elevated blood pressure, acute kidney injury, chronic kidney disease, plasma transfusion, and emergency surgery to be risk factors associated with TACO.45 Also, a recent retrospective cohort study of 66 TACO patients found cardiac failure, renal failure, and the degree of positive fluid balance as risk factors for TACO development.46 Previously, congestive heart failure and renal dysfunction were identified as common features in TACO in a retrospective study of 98 TACO patients, which, in addition, also identified an age of over 70 years as a risk factor for TACO development.47 Advanced age was also shown to increase the incidence of TACO in perioperative noncardiac surgery patients.48 The second hit in TACO may be reflected by suboptimal fluid management and inappropriate infusion practices (such as rapid infusion rates), which have frequently been related to the onset of TACO.47 In a perioperative setting of noncardiac surgery, the incidence of TACO was found to be increased with the volume transfused and the total intraoperative fluid balance.48 Remarkably, it was recently observed that the degree of positive fluid balance appeared to be associated less with the development of TACO than with the development of circulatory overload in the absence of transfusion.46 This lower association of positive fluid balance with the development of TACO may be due to the increase in colloid-osmotic pressure due to the transfusion product stimulating fluids from the extravascular space into the intravascular space in an attempt to contribute a more effective circulating volume. Alternatively, it may indicate that other factors in the transfused blood product besides the transfusion volume could play a role in the onset of TACO. Combined with the first hit, these factors may possibly contribute to inflammation in the transfused recipient resulting in TACO, as will be discussed later.
In TRALI, first-hit risk factors include chronic alcohol abuse, shock, liver surgery, current smoking, higher peak airway pressure while undergoing mechanical ventilation, positive intravascular fluid balance,49 low interleukin-10 (IL-10) levels,50-53 and systemic inflammation.53 Systemic inflammation may be reflected in the plasma cytokine profiles (increased IL-6 and IL-8 levels as discussed later) but also via increased levels of C-reactive protein (CRP). CRP is an acute-phase protein that rapidly increases during acute infections and inflammation and is widely used in clinic as an inflammatory biomarker.54 CRP was shown to be elevated in TRALI patients55 and functionally enabled the first hit in the development of TRALI in a murine model by increasing the levels of the neutrophil (PMN)-chemoattractant macrophage inflammatory protein (MIP)-2 (murine homolog of IL-8), resulting in increased pulmonary PMN accumulation.56 The second hit in TRALI may be conveyed by antileukocyte antibodies or other factors present in the transfusion product. In ∼80% of cases, anti-HLA class I or II or anti-HNA antibodies are implicated to be involved in triggering TRALI,41 although this may be even higher depending on the detection methods used.57 In the remaining 20% of TRALI cases, non–antibody factors/biological response modifiers are suggested to contribute the second hit, and these may possibly include lipid mediators, extracellular vesicles, and aged blood cells (as described in “Non-antibody–mediated TRALI”).58
Cytokine profiles
Cytokine profiles may shed light on the pathophysiologic environment of TACO and TRALI; however, in TACO, this has rarely been investigated. A nested case control study involving 29 TACO patients, 147 control patients, as well as 70 TRALI patients has initiated these important investigations.50 In this study, the proinflammatory cytokine IL-6 was found to be significantly elevated in posttransfusion TACO patients compared with matched controls. Levels of the proinflammatory cytokine IL-8, on the other hand, were not elevated in pre- or posttransfusion TACO patients, whereas in TRALI patients, both IL-6 and IL-8 levels were elevated pre- as well as posttransfusion in the same study.50 Furthermore, the anti-inflammatory cytokine IL-10 was found to be increased in both pre- and posttransfusion TACO patients, whereas IL-10 levels remained low in pre- and posttransfusion TRALI patients.50 No changes were found in pre- or posttransfusion TACO or TRALI patients regarding granulocyte macrophage-colony stimulating factor (GM-CSF) or tumor necrosis factor-α (TNF-α).50 Other studies examining cytokine levels in TRALI have reported increased levels of pre- and posttransfusion IL-6 and IL-8 in cardiac surgery patients,59 increased levels of pretransfusion IL-8,49 and low/nonelevated levels of posttransfusion IL-10.51 One study, however, found IL-10 levels to be increased in TRALI patients.60 This may be due to the fact that this study investigated fold changes of paired samples prior to and following transfusion, in contrast to the comparison of posttransfusion TRALI samples vs transfused controls.50,51 The posttransfusion cytokine levels in TACO and TRALI based on these studies are summarized in Table 3. The data so far appear promising, especially considering that they may facilitate the challenging distinction between TACO and TRALI (in a symptomatic setting posttransfusion). Low IL-8 levels in combination with elevated IL-10 levels may potentially help support a diagnosis of TACO, whereas increased IL-8 levels in combination with low IL-10 levels may potentially aid in the diagnosis of TRALI. It will be very important, however, to validate these cytokine profiles in larger patient cohorts of TRALI and especially TACO.
Pathophysiological mechanisms
TACO
Research into the pathophysiology of TACO has been surprisingly limited over the years.7 The pathophysiology of TACO was initially thought to be identical to the well-studied mechanisms of congestive heart failure or pulmonary hydrostatic edema, but is now recognized to be more complex. It remains unknown which pathways are involved in generating the pulmonary hydrostatic edema (protein-poor edema fluid) in TACO. Recently, studies have started to investigate risk factors for development of TACO and, as previously discussed, cardiac failure, renal failure, and positive fluid balance are first-hit components responsible for the poor adaptability for volume overload. Second hits may be suboptimal fluid management or other components present in the transfusion product. Secretion of IL-10, which was found to be increased in TACO patients,50 is known to require specific stimulation such as by microbial products or specific antibodies.52,61 It is therefore possible that factors in the transfused blood product could perhaps be mediating this effect. Regarding IL-10 levels in TACO, however, an important role for the recipient’s status (comorbidities or postoperative status) must be considered as well. IL-10 levels were also found to be increased in pretransfusion TACO patients, although apparently to a lower extent than in posttransfusion TACO patients.50 Furthermore, 1 study found that the occurrence of TACO decreased ∼50% by the introduction of universal leukoreduced products,62 indicating that other factors besides volume overload may be involved. As this was only a single retrospective study, further validation in animal models, in prospective clinical trials, and via hemovigilance systems is required to investigate whether TACO rates indeed decrease due to leukoreduction. Similarly, it has been observed that in perioperative noncardiac patients, the rates of TACO incidence decreased from 2004 to 2011,48 which may be related to the exclusion of female plasma donors for transfusions in this period. Alternatively, it may be well possible that this decrease in TACO incidence could also have been related to changes in clinical practice (such as declines in transfusion related to patient blood management). Other factors that could potentially be involved may be related to inflammation or cytokine levels. The proinflammatory cytokine IL-6 was found to be increased in TACO patients,50 and fever was described to occur in one-third of the patients developing TACO.36 It will be important to further research the contribution of the transfusion product beyond the volume alone, including the role of inflammation. Because of lack of experimental animal models for TACO, almost nothing is known about the cellular moieties involved in the pathophysiology of TACO. One paper reported the presence of intra-alveolar neutrophils (without the formation of neutrophil extracellular traps [NETs]) in the lungs of a TACO patient,63 which additionally supports the involvement of an inflammatory component in TACO. The current knowledge regarding the pathophysiology of TACO is depicted in Figure 2.
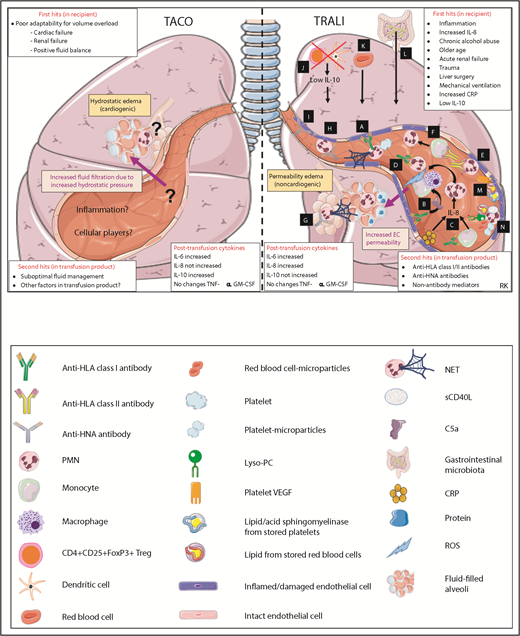
Pathophysiological mechanisms of TACO and TRALI in the lungs. Pathways A-N are systematically discussed in the main text. sCD40L, soluble CD40 ligand; VEGF, vascular endothelial growth factor. This figure was in part created with images adapted from Servier Medical Art by Servier, which is licensed under a Creative Commons Attribution 3.0 Unported License.
Pathophysiological mechanisms of TACO and TRALI in the lungs. Pathways A-N are systematically discussed in the main text. sCD40L, soluble CD40 ligand; VEGF, vascular endothelial growth factor. This figure was in part created with images adapted from Servier Medical Art by Servier, which is licensed under a Creative Commons Attribution 3.0 Unported License.
TRALI
Although much more is known regarding the pathophysiology of TRALI as compared with TACO, it remains complex and still incompletely understood. TRALI results in noncardiogenic permeability edema (protein-rich edema fluid). Most of the data on the TRALI pathophysiological mechanisms are derived from animal models or from in vitro cultures of human lung endothelial cells. Animal models of TRALI have significantly facilitated research into the TRALI pathophysiology,64 but controversies have arisen. The complexity comes from the use of various animal models, various triggers (different types of antibodies or biological response modifiers), and different experimental conditions. When assessing cellular involvement in the pathophysiology of TRALI, which is summarized in Table 4, it appears that PMNs are the key pathogenic cells that mediate lung damage in TRALI,65 whereas CD4+CD25+FoxP3+ T-regulatory (Treg) cells and dendritic cells appear to be the key protective cells.52 Furthermore, it seems that monocytes and/or macrophages and red blood cells convey a pathogenic role in TRALI (Table 4). As further described in Table 4, the role of platelets appears pathogenic in TRALI; however, reports have also strongly argued against this pathogenic involvement. Similarly, CD8+ T lymphocytes and B lymphocytes do not appear to have a significant role in the pathophysiology of TRALI (Table 4). Suggested TRALI pathophysiologic mechanisms also depend on the type of antibodies (anti-HLA class I, anti-HLA class II, or anti-HNA antibodies) or biological response modifiers (such as lipids from stored platelets or red blood cells) involved.
Overview of evidence for and against the involvement of specific cell types in the pathogenesis of TRALI, based on both TRALI animal models and/or human data
| Cell type | Supporting evidence for involvement in TRALI | Evidence against involvement in TRALI | Overall effect in TRALI |
|---|---|---|---|
| PMNs | • PMNs are observed in TRALI lungs upon autopsy86,87 • In vitro TRALI models have shown PMN-induced endothelial cell damage. Antibody-mediated second hits: anti-HNA-3a antibody,74 anti-HLA antibody,70 low IgM serum.97 Non-antibody–mediated second hits: lyso-PC,98 soluble CD40 ligand,85 platelet microparticles82 • NETs detected in plasma (n = 14) and lungs of TRALI patients and in murine plasma and lung microcirculation of mice undergoing anti-MHC class I antibody–mediated TRALI (upon LPS priming)63 Suggested mechanism: Platelets induce NET formation and NETs induce lung injury via direct toxicity to pulmonary endothelial cells • NET biomarkers found in serum of TRALI patients (n = 5) and in murine lung alveoli using an anti-MHC class I antibody–mediated TRALI model (upon LPS priming)73 Suggested mechanism: NETs formed through direct priming of PMNs by anti-HNA-3a antibodies73 • PMNs were shown to interact with von Willebrand factor via CTL-2, enabling anti-HNA-3a to signal via CD11b/CD18, resulting in PMN-activation and agglutination99 • Pulmonary PMN infiltration has frequently been observed in multiple animal models of TRALI, both antibody mediated52,56,63,66,68,75,76,99-103 and non-antibody mediated103,104 • PMN-FcγRs were found to be essential in a murine anti-MHC class I antibody–mediated TRALI model (without LPS priming)66 Suggested mechanism: Anti-MHC class I antibody binds to the pulmonary endothelium and sequesters PMNs via their FcγRs, resulting in PMN activation and TRALI induction66 • PMN depletion fully prevented TRALI occurrence using murine anti-MHC class I antibody–mediated TRALI models (without LPS priming)52,66,100 • PMN and ROS are critically required for TRALI induction in a murine model of anti-MHC class I antibody–mediated TRALI (model based on CD4+ T-cell depletion and without LPS, using C57BL/6 gp91phox knock-out mice)52 • In a 2-event in vivo rat TRALI model, inflammatory priming PMNs induced MHC class II surface expression and pulmonary endothelium activation. Anti-MHC class II antibodies subsequently targeted sequestered PMNs resulting in TRALI71 | • TRALI reported in a neutropenic patient105 • No alveolar PMN influx observed upon lung tissue histology analysis of 2 TRALI patients106 • Occurrence of TRALI after PMN depletion in a murine anti-HNA-3a TRALI model (despite a decreased disease severity)75 • No dependence on PMNs (and partial independence of FcγRs) but on complement component C5a in a murine anti-MHC class I antibody–mediated TRALI model67 | Pathogenic |
| Monocytes and/or macrophages | • Anti-MHC class I antibody–mediated murine (SCID mice, without LPS priming) TRALI induction was completely abrogated upon monocyte depletion (and restored upon repletion with purified monocytes)68 Suggested mechanism: Anti-MHC class I antibody binding to monocytes induces the secretion of the PMN-chemoattractant MIP-2 (murine homolog of IL-8), resulting in pulmonary PMN recruitment and TRALI68 • Depletion or inactivation of monocytes/macrophages in vivo fully suppressed TRALI in murine model of anti-MHC class I antibody–mediated TRALI (without LPS priming)67 Suggested mechanism: Anti-MHC class I antibody binding to endothelial cells activates complement with production of C5a, which then binds to the C5aR on monocytes/macrophages, attracting these cells to the lungs and inducing them to produce ROS, damaging the endothelium and inducing TRALI67 • Anti-HLA class II antibodies induce monocyte activation in an ex vivo rodent model of TRALI, which subsequently results in activation of PMNs, which then damage the pulmonary endothelium72 • TRALI monocytes were activated in vitro by sera implicated in TRALI, resulting in increased production of TNF-α, IL-1β, and Tissue Factor over a 4-h period compared with control sera107 | Pathogenic | |
| Platelets | • Occurrence of thrombocytopenia in TRALI patients37-40 • Reported thrombocytopenia in murine anti-MHC class I antibody–mediated TRALI model based on CRP infusion56 • Platelet depletion (platelet depleting antibody injected 4 h prior to TRALI induction) protected mice from TRALI in a murine anti-MHC class I–mediated TRALI model (with LPS priming)100 Suggested mechanism: Platelets induce NET formation (which induce lung injury via direct toxicity to pulmonary endothelial cells) and targeting platelet activation with aspirin or a glycoprotein IIb/IIIa inhibitor decreased NET formation and TRALI63 • Stored platelet-derived vascular endothelial growth factor potentially mediates increased lung vascular permeability in isolated-perfused rat lungs79 • Plasma and lipids from stored platelets enable TRALI in an LPS-primed rat model78 • Lyso-PC from stored platelets causes pulmonary and systemic coagulopathy in an LPS-primed rat model, via PMN-priming in vitro81 • Platelet-derived microparticles enable PMN-mediated pulmonary endothelial cell damage in vitro82 • Aged platelets induce lung injury via acid sphingomyelinase in an LPS-primed murine TRALI model80 | • No thrombocytopenia detected in murine model of anti-MHC class I antibody–mediated TRALI (without LPS priming)66 • No effect of inducing thrombocytopenia or of pharmacological and genetic targeting of platelet functions, on the development of TRALI in a murine anti-MHC class I antibody–mediated TRALI model (with LPS priming)108 • Platelet depletion (with platelet depleting antibody injected 24 and 48 h prior to TRALI induction or by injection of neuraminidase) did not suppress TRALI development in a murine model of anti-MHC class I–mediated TRALI (without LPS priming)67 | Pathogenic? Dispensable? |
| Red blood cells | • Plasma and lipids from red blood cells and leukoreduced red blood cells primed PMNs and induced ALI in rat model with LPS priming84 • Red blood cell–derived microparticles may prime PMNs and enable TRALI in LPS-primed mice83 • Lipids and supernatants from stored red blood cells activate pulmonary endothelium through the BLT2 receptor and protein kinase C activation, and predispose to ALI77 • Transfusion of supernatant of aged red blood cells induced lung inflammation and coagulopathy in LPS-primed rats109 • Injection of anti–red blood cell antibody 1 d before induction of anti-MHC class I antibody–mediated murine TRALI resulted in total suppression of TRALI development in BALB/c mice67 | • Transfusion of 35-d stored autologous red blood cells in LPS-primed human volunteers (with confirmed sepsis/endotoxemia) did not result in lung injury89 | Pathogenic |
| CD4+CD25+FoxP3 Tregs and dendritic cells | • Depletion of CD4+CD25+FoxP3+ Tregs or of CD11c+ dendritic cells resulted in low IL-10 levels, which enabled anti-MHC class I antibody–mediated murine TRALI (protection against anti-MHC class I antibody–mediated TRALI was associated with increased IL-10 levels)52 Suggested mechanism: Depletion of CD4+CD25+FoxP3+ Tregs or of CD11c+ dendritic cells in mice results in low IL-10 levels, which enables increased anti-MHC class I antibody–mediated plasma MIP-2 levels (murine homolog of IL-8), pulmonary PMN infiltration, ROS production, and TRALI development with impaired lung function52 | Protective | |
| CD8+ T cells | Depletion of CD8+ T cells did not significantly result in the onset of anti-MHC class I antibody–mediated murine TRALI52 | No significant involvement | |
| B cells | Depletion of B cells did not significantly result in the onset of anti-MHC class I antibody–mediated murine TRALI52 | No significant involvement |
| Cell type | Supporting evidence for involvement in TRALI | Evidence against involvement in TRALI | Overall effect in TRALI |
|---|---|---|---|
| PMNs | • PMNs are observed in TRALI lungs upon autopsy86,87 • In vitro TRALI models have shown PMN-induced endothelial cell damage. Antibody-mediated second hits: anti-HNA-3a antibody,74 anti-HLA antibody,70 low IgM serum.97 Non-antibody–mediated second hits: lyso-PC,98 soluble CD40 ligand,85 platelet microparticles82 • NETs detected in plasma (n = 14) and lungs of TRALI patients and in murine plasma and lung microcirculation of mice undergoing anti-MHC class I antibody–mediated TRALI (upon LPS priming)63 Suggested mechanism: Platelets induce NET formation and NETs induce lung injury via direct toxicity to pulmonary endothelial cells • NET biomarkers found in serum of TRALI patients (n = 5) and in murine lung alveoli using an anti-MHC class I antibody–mediated TRALI model (upon LPS priming)73 Suggested mechanism: NETs formed through direct priming of PMNs by anti-HNA-3a antibodies73 • PMNs were shown to interact with von Willebrand factor via CTL-2, enabling anti-HNA-3a to signal via CD11b/CD18, resulting in PMN-activation and agglutination99 • Pulmonary PMN infiltration has frequently been observed in multiple animal models of TRALI, both antibody mediated52,56,63,66,68,75,76,99-103 and non-antibody mediated103,104 • PMN-FcγRs were found to be essential in a murine anti-MHC class I antibody–mediated TRALI model (without LPS priming)66 Suggested mechanism: Anti-MHC class I antibody binds to the pulmonary endothelium and sequesters PMNs via their FcγRs, resulting in PMN activation and TRALI induction66 • PMN depletion fully prevented TRALI occurrence using murine anti-MHC class I antibody–mediated TRALI models (without LPS priming)52,66,100 • PMN and ROS are critically required for TRALI induction in a murine model of anti-MHC class I antibody–mediated TRALI (model based on CD4+ T-cell depletion and without LPS, using C57BL/6 gp91phox knock-out mice)52 • In a 2-event in vivo rat TRALI model, inflammatory priming PMNs induced MHC class II surface expression and pulmonary endothelium activation. Anti-MHC class II antibodies subsequently targeted sequestered PMNs resulting in TRALI71 | • TRALI reported in a neutropenic patient105 • No alveolar PMN influx observed upon lung tissue histology analysis of 2 TRALI patients106 • Occurrence of TRALI after PMN depletion in a murine anti-HNA-3a TRALI model (despite a decreased disease severity)75 • No dependence on PMNs (and partial independence of FcγRs) but on complement component C5a in a murine anti-MHC class I antibody–mediated TRALI model67 | Pathogenic |
| Monocytes and/or macrophages | • Anti-MHC class I antibody–mediated murine (SCID mice, without LPS priming) TRALI induction was completely abrogated upon monocyte depletion (and restored upon repletion with purified monocytes)68 Suggested mechanism: Anti-MHC class I antibody binding to monocytes induces the secretion of the PMN-chemoattractant MIP-2 (murine homolog of IL-8), resulting in pulmonary PMN recruitment and TRALI68 • Depletion or inactivation of monocytes/macrophages in vivo fully suppressed TRALI in murine model of anti-MHC class I antibody–mediated TRALI (without LPS priming)67 Suggested mechanism: Anti-MHC class I antibody binding to endothelial cells activates complement with production of C5a, which then binds to the C5aR on monocytes/macrophages, attracting these cells to the lungs and inducing them to produce ROS, damaging the endothelium and inducing TRALI67 • Anti-HLA class II antibodies induce monocyte activation in an ex vivo rodent model of TRALI, which subsequently results in activation of PMNs, which then damage the pulmonary endothelium72 • TRALI monocytes were activated in vitro by sera implicated in TRALI, resulting in increased production of TNF-α, IL-1β, and Tissue Factor over a 4-h period compared with control sera107 | Pathogenic | |
| Platelets | • Occurrence of thrombocytopenia in TRALI patients37-40 • Reported thrombocytopenia in murine anti-MHC class I antibody–mediated TRALI model based on CRP infusion56 • Platelet depletion (platelet depleting antibody injected 4 h prior to TRALI induction) protected mice from TRALI in a murine anti-MHC class I–mediated TRALI model (with LPS priming)100 Suggested mechanism: Platelets induce NET formation (which induce lung injury via direct toxicity to pulmonary endothelial cells) and targeting platelet activation with aspirin or a glycoprotein IIb/IIIa inhibitor decreased NET formation and TRALI63 • Stored platelet-derived vascular endothelial growth factor potentially mediates increased lung vascular permeability in isolated-perfused rat lungs79 • Plasma and lipids from stored platelets enable TRALI in an LPS-primed rat model78 • Lyso-PC from stored platelets causes pulmonary and systemic coagulopathy in an LPS-primed rat model, via PMN-priming in vitro81 • Platelet-derived microparticles enable PMN-mediated pulmonary endothelial cell damage in vitro82 • Aged platelets induce lung injury via acid sphingomyelinase in an LPS-primed murine TRALI model80 | • No thrombocytopenia detected in murine model of anti-MHC class I antibody–mediated TRALI (without LPS priming)66 • No effect of inducing thrombocytopenia or of pharmacological and genetic targeting of platelet functions, on the development of TRALI in a murine anti-MHC class I antibody–mediated TRALI model (with LPS priming)108 • Platelet depletion (with platelet depleting antibody injected 24 and 48 h prior to TRALI induction or by injection of neuraminidase) did not suppress TRALI development in a murine model of anti-MHC class I–mediated TRALI (without LPS priming)67 | Pathogenic? Dispensable? |
| Red blood cells | • Plasma and lipids from red blood cells and leukoreduced red blood cells primed PMNs and induced ALI in rat model with LPS priming84 • Red blood cell–derived microparticles may prime PMNs and enable TRALI in LPS-primed mice83 • Lipids and supernatants from stored red blood cells activate pulmonary endothelium through the BLT2 receptor and protein kinase C activation, and predispose to ALI77 • Transfusion of supernatant of aged red blood cells induced lung inflammation and coagulopathy in LPS-primed rats109 • Injection of anti–red blood cell antibody 1 d before induction of anti-MHC class I antibody–mediated murine TRALI resulted in total suppression of TRALI development in BALB/c mice67 | • Transfusion of 35-d stored autologous red blood cells in LPS-primed human volunteers (with confirmed sepsis/endotoxemia) did not result in lung injury89 | Pathogenic |
| CD4+CD25+FoxP3 Tregs and dendritic cells | • Depletion of CD4+CD25+FoxP3+ Tregs or of CD11c+ dendritic cells resulted in low IL-10 levels, which enabled anti-MHC class I antibody–mediated murine TRALI (protection against anti-MHC class I antibody–mediated TRALI was associated with increased IL-10 levels)52 Suggested mechanism: Depletion of CD4+CD25+FoxP3+ Tregs or of CD11c+ dendritic cells in mice results in low IL-10 levels, which enables increased anti-MHC class I antibody–mediated plasma MIP-2 levels (murine homolog of IL-8), pulmonary PMN infiltration, ROS production, and TRALI development with impaired lung function52 | Protective | |
| CD8+ T cells | Depletion of CD8+ T cells did not significantly result in the onset of anti-MHC class I antibody–mediated murine TRALI52 | No significant involvement | |
| B cells | Depletion of B cells did not significantly result in the onset of anti-MHC class I antibody–mediated murine TRALI52 | No significant involvement |
Antibody-mediated TRALI
Antibody-mediated mechanisms of TRALI are depicted in pathways A-L (Figure 2), and non-antibody–mediated mechanisms are illustrated in pathways M and N of Figure 2. Using an anti–major histocompatibility complex (MHC) class I antibody–mediated murine model, it was suggested that the anti-MHC class I antibody binds to the pulmonary endothelium and sequesters PMNs via their FcγRs, resulting in PMN activation.66 Subsequently, it has been described that platelets may induce NET formation, causing the NETs to induce direct toxicity to the pulmonary endothelium enabling TRALI63 (Figure 2 pathway A). Another mechanism may be that anti-MHC class I antibodies may bind the pulmonary endothelial cells activating the complement cascade, resulting in the production of C5a.67 C5a may then bind to the C5a receptor on monocytes/macrophages, attracting these cells to the lungs and inducing them to produce reactive oxygen species (ROS), which then damages the pulmonary endothelium, resulting in TRALI67 (Figure 2 pathway B). Importantly, ROS was shown to be critically required for anti-MHC class I antibody–meditated murine TRALI as was demonstrated using gp91phox knock-out mice.52 Anti-MHC class I antibodies may also target monocytes, inducing secretion of MIP-2, resulting in pulmonary PMN recruitment and TRALI.68 CRP together with anti-MHC class I antibody can also cause a synergistic increase in both MIP-2 levels and pulmonary PMN accumulation, enabling TRALI.56 CRP may also directly target the endothelium,69 and CRP has been described to be elevated in TRALI patients55 (Figure 2 pathway C). Anti-HLA-A2 antibodies were also shown to activate PMNs and induce endothelial cell damage in vitro70 (Figure 2 pathway D). Inflammatory priming was shown to induce MHC class II surface expression on PMNs and activation of the pulmonary endothelium, enabling anti-MHC class II antibodies to directly target sequestered PMNs inducing TRALI in a rat model71 (Figure 2 pathway E). Furthermore, anti-HLA class II antibodies were shown to induce monocyte activation in an ex vivo rodent TRALI model, which subsequently resulted in PMN activation that then damaged the pulmonary endothelium causing TRALI72 (Figure 2 pathway F). Anti–HNA-3a antibodies were shown to directly prime PMNs, resulting in the formation of NETs73 (Figure 2 pathway G) as well as to cause PMN priming enabling PMN-mediated pulmonary endothelial cell damage in vitro74 (Figure 2 pathway H). Alternatively, anti–HNA-3a antibodies have been shown to directly target lung endothelium cells, causing barrier dysfunction, although the presence of PMNs did aggravate this endothelial barrier dysfunction75 (Figure 2 pathway I). Using an anti-MHC class I–mediated murine TRALI model, CD4+CD25+FoxP3+ Tregs and dendritic cells were found to be key protecting cells in TRALI via upregulation of IL-10.52 This was demonstrated by in vivo Treg and dendritic cell depletions, which corresponded to low IL-10 levels and subsequently enabled anti-MHC class I antibody–mediated TRALI. In addition, this was verified by using IL-10 knock-out mice, which were shown to be hypersensitive to anti-MHC class I antibody–mediated murine TRALI induction52 (Figure 2 pathway J). Red blood cells may also be pathogenic in antibody-mediated TRALI because RBC depletion with injection with an anti-RBC antibody prevented the onset of anti-MHC class I antibody–mediated TRALI in BALB/c mice67 (Figure 2 pathway K). The gastrointestinal microbiota was also shown to contribute to the development of antibody-mediated murine TRALI.76 Mice housed in a more sterile environment (specific pathogen-free mice) were shown to be resistant to anti-MHC class I–mediated TRALI, whereas less sterile housed mice (barrier-free mice) were sensitive to anti-MHC class I antibody–mediated TRALI but become resistant to TRALI upon gut flora depletion using broad-spectrum antibiotics.76 Interestingly, fecal transplants from TRALI-susceptible mice into the sterile-housed TRALI-resistant mice were able to restore the TRALI responses.76 The exact interplay between the gastrointestinal flora and TRALI induction mechanisms will need to be further investigated (Figure 2 pathway L).
Non-antibody–mediated TRALI
Non-antibody–mediated mechanisms of TRALI may include direct targeting of biological response modifiers toward the pulmonary endothelium. Lipids from stored red blood cells may activate the pulmonary endothelium via the BLT2 receptor and protein kinase C predisposing to acute lung injury.77 In addition, plasma and lipids from stored platelets may enable TRALI in an lipopolysaccharide (LPS)-primed rat model,78 and stored platelet-derived vascular endothelial growth factor may also potentially increase lung vascular permeability in rat lungs.79 Acid sphingomyelinase from aged platelets has also been shown to induce TRALI in LPS-primed mice80 (Figure 2 pathway M). Biological response modifiers may also mediate TRALI via PMNs, such as lyso-PC, from stored platelets, which may cause pulmonary and systemic coagulopathy in LPS-primed mice in vitro via PMN priming.81 Furthermore, both platelet- and red blood cell microparticles have been demonstrated to prime PMNs and mediate TRALI in human pulmonary microvascular endothelial cells in vitro and in mice, respectively.82,83 In addition, lipids from red blood cells were shown to prime PMNs and induce acute lung injury in LPS-primed mice.84 Soluble CD40 ligand (sCD40L) has been demonstrated to accumulate in stored blood components and enabled PMN-mediated endothelial cell damage in vitro85 (Figure 2 pathway N). Notably, regarding the role of PMNs in the 2-hit TRALI models, these cells may differentially accumulate in the lungs at various stages between the first and second hits of TRALI induction (Figure 2 pathway C, E, J, or N).
Implications for human TRALI disease
The data supporting these mechanisms in human cases of TRALI are limited. Supporting data that are derived from TRALI patients directly include the discussed plasma cytokine profiles (see previous section), increased levels of CRP,55 detection of NETs in plasma and lungs of TRALI patients,63 presence of NET biomarkers in serum of TRALI patients,73 and the observation of PMNs in TRALI lungs upon autopsy.86,87 Notably, the suggested mechanisms of non-antibody–mediated TRALI have not been confirmed to occur in human studies.88,89
Potential therapies and future directions
Unfortunately, for both TACO and TRALI, only supportive measures are available, and specific therapies are lacking. Supportive measures for TACO may include diuresis, oxygen, and intubation. For TRALI, supportive measures may include oxygen, intubation, and judicious fluid and pressor management to maintain hemodynamics. Preventive strategies for TRALI (TRALI mitigation) include donor deferral based on antibody screening (anti-HNA and anti-HLA antibodies), donor deferral based on history of pregnancy or history of transfusion, and deferral of all female donors (use of male-only plasma donors).90 These TRALI mitigation strategies have been successful in significantly reducing the incidence of TRALI. There was a reduction in plasma-induced TRALI cases in at-risk patient populations (surgery, intensive care unit) and a decreased tendency of plasma-induced TRALI cases in a general patient population.90 In addition, the FDA reported 35 TRALI cases in 2006, and upon TRALI mitigation, this was reduced to 8 cases in 2016.15 The SHOT report also described a strong decline from 24 cases in 2003 that dropped to 3 cases in 2017 after introduction of TRALI mitigation strategies.14 Furthermore, using prospective, active surveillance, Toy et al observed a TRALI incidence of 2.57 (95% confidence interval, 1.72-3.86) in 2006 vs 0.81 (95% confidence interval, 0.44-1.49) in 2009 per 10 000 transfused units (P = .002).49 Similarly, the incidence of TACO in perioperative noncardiac surgery patients was observed to be 5.5% (119/2162) in 2004 vs 3.0% (57/1908) in 2011 (P < .001).48 In the same patient population, however, the incidence of TRALI/possible TRALI was 1.3% (23/1613) in 2004 vs 1.4% (22/1562) in 2011 (P = .72), indicating that no decline in the incidence of TRALI/possible TRALI occurred despite the introduction of TRALI mitigation strategies.91 The French Haemovigilance Network did also not observe a significant reduction in the overall incidence of TRALI cases between 2007 and 2013.92 It may be possible, however, that the inclusion of possible TRALI cases together with TRALI cases in these studies could have obscured a decline in true TRALI cases due to the mitigation measures. Limitations of the TRALI mitigation strategies include a shortage of male AB plasma93 and no decrease in red blood cell and platelet transfusion-related TRALI.94 Furthermore, no significant effect on 30-day TRALI mortality was observed with low-risk TRALI donor strategies in plasma-induced TRALI cases.90 It is therefore important to pursue the identification of potential therapies for TRALI.
Recently, potential therapies for TRALI have been reviewed,53 and it was concluded that the most promising therapeutic strategies to explore are IL-10 therapy, downregulation of CRP levels, targeting ROS, or blocking IL-8 receptors, all focused on the transfusion recipient.53 More validation and confirmation will be required first for other therapies focused on the transfusion recipient or for therapeutic interventions aimed at the blood transfusion product.53 IL-10 therapy was shown to not only prophylactically but also therapeutically (ie, after onset of TRALI symptoms) prevent and rescue TRALI in a murine model.52 Because IL-10 levels are not elevated in posttransfusion TRALI patients,50,51 IL-10 therapy may be particularly interesting to explore as a potential therapy. IL-10 administration to healthy volunteers has been shown to be safe and well tolerated.95 Caution, however, is strongly advised because IL-10 administration may impair the host resistance to infectious pathogens in critically ill patients. We have summarized the potential TRALI therapies in Figure 3.
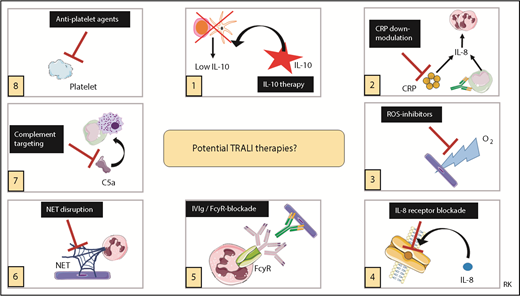
Potential TRALI therapies. Summary of potential therapeutic targets in TRALI. IVIg, IV immunoglobulin. This figure was in part created with images adapted from Servier Medical Art by Servier, which is licensed under a Creative Commons Attribution 3.0 Unported License.
Potential TRALI therapies. Summary of potential therapeutic targets in TRALI. IVIg, IV immunoglobulin. This figure was in part created with images adapted from Servier Medical Art by Servier, which is licensed under a Creative Commons Attribution 3.0 Unported License.
Regarding potential therapies for TACO, much more pathophysiological knowledge will need to be required first, and development of animal models will be important. It will be imperative to investigate if patients at risk for TACO could be better identified pretransfusion by improving the screening for occult cardiac insufficiency (eg, using echocardiography or measurement of N-terminal pro-BNP). In addition, the effect of reducing the rate and volume of transfusions should be researched in patients at risk for TACO development.
Overall, TACO and TRALI remain significant clinical problems. Further research into the pathophysiological mechanisms will be critical for improvement of diagnostic approaches and for the design and assessment of potential therapeutic strategies. It will be important to expand on biomarker investigations and to validate risk factors for TRALI and TACO. The development of TACO animal models is highly warranted to dissect the disease pathology because other factors besides volume appear to play a significant role in TACO. These approaches may significantly contribute in combating these life-threatening complications of blood transfusion.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from Lund University (J.W.S.), Crafoordska Stiftelsen 20170829 (J.W.S.), Vetenskapsrådet (Swedish Research Council, VR) 2017-01779 (J.W.S.), and Avtal om Läkarutbildning och Forskning (J.W.S.). R.K. was the recipient of a young investigator award by the Royal Physiographic Society of Lund.
Authorship
Contribution: J.W.S. wrote and edited the manuscript; J.R. edited the manuscript; and R.K. wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for R.K. is Department of Experimental Immunohematology, Sanquin Research, and Landsteiner Laboratory, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands.
Correspondence: John W. Semple Lund University, BMC C14, Klinikgatan 26, 221 84 Lund, Sweden; e-mail: john_w.semple@med.lu.se; and Rick Kapur, Department of Experimental Immunohematology, Sanquin Research, and Landsteiner Laboratory, Amsterdam UMC, University of Amsterdam, Plesmanlaan 125, 1066 CX Amsterdam, The Netherlands; e-mail: r.kapur@sanquin.nl.
