

In this issue of Blood, have identified a novel regulatory mechanism that ensures proper splicing of human α-globin pre-messenger RNA (pre-mRNA).1
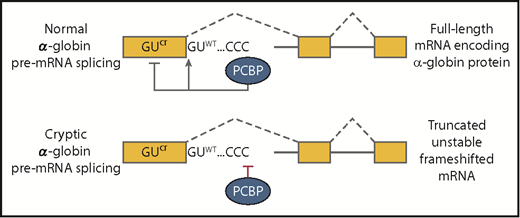
Top panel shows that normal α-globin splicing is favored and cryptic splicing is suppressed when polyC-binding proteins are bound at the C-rich element downstream of exon 1. Proper splicing generates a full-length mRNA that encodes α-globin. The bottom panel depicts cryptic splicing that frequently occurs when polyC-binding proteins fail to bind. The resulting transcript cannot encode α-globin because the truncation of exon 1 will alter the translational reading frame and induce nonsense-mediated decay of the RNA. CCC, C-rich sequence; GUcr, cryptic splice site in exon 1; GUWT, normal splice site at the boundary between exon 1 and intron 1.
Top panel shows that normal α-globin splicing is favored and cryptic splicing is suppressed when polyC-binding proteins are bound at the C-rich element downstream of exon 1. Proper splicing generates a full-length mRNA that encodes α-globin. The bottom panel depicts cryptic splicing that frequently occurs when polyC-binding proteins fail to bind. The resulting transcript cannot encode α-globin because the truncation of exon 1 will alter the translational reading frame and induce nonsense-mediated decay of the RNA. CCC, C-rich sequence; GUcr, cryptic splice site in exon 1; GUWT, normal splice site at the boundary between exon 1 and intron 1.
One could be forgiven for thinking that splicing of this well-studied gene is fully understood, but Ji et al have described an essential new element: a C-rich sequence located a few nucleotides downstream of the intron 1 splice donor site that favors proper splicing while suppressing use of a competing cryptic splice site in the exon (see figure). Moreover, they report that polyC-binding proteins must interact with this intronic element to enhance proper splicing. If the C-rich element is mutated or expression of the polyC-binding proteins is reduced, then splice site choice shifts dramatically to a cryptic splice donor site in the coding region of exon 1. Splicing at the cryptic site truncates the exon and alters the translational reading frame, which renders the aberrant mRNA incapable of encoding α-globin protein. Interestingly, a naturally occurring mutation at the correct splice donor site had a similar splicing phenotype in an α-thalassemia patient (ie, splicing shifted to the cryptic site to produce a nonfunctional mRNA).2 Together these results show that the cryptic splice site is perfectly functional when not in competition with the correct splice site. However, normal cells almost entirely suppress the splicing at the cryptic site by the polyC-binding protein-mediated mechanism.
PolyC binding proteins are encoded by 2 genes, PCBP1 and PCBP2. They are highly expressed in erythroid cells and have several documented roles to promote proper erythroid gene expression. Via binding to the 3′ untranslated region of α-globin transcripts, polyC-binding proteins can stimulate cleavage and the polyadenylation of nascent transcripts and can also stabilize mature cytoplasmic α-globin mRNA against degradation.3 The same proteins can enhance splicing of selected alternative exons via binding to RNA at C-rich elements in the polypyrimidine tracts upstream of their cognate splice acceptor sites.4 In that context they interact with key splicing components, including U2 small nuclear ribonucleoprotein (snRNP) and the canonical polypyrimidine tract–binding protein U2AF65, acting to recruit them to the splice acceptor site. In their study, Ji et al demonstrate a new function for polyC-binding proteins at splice donor sites, where they presumably act to enhance assembly of U1 snRNP particles at 5′ splice donor sites.
These findings serve as a reminder that accurate selection of authentic splice sites is a complex challenge for the splicing machinery. Despite the lack of a nucleus in mature red cells, regulation of splicing is just as important during terminal erythropoiesis as it is in other cell types (reviewed in Conboy5 ). The root of the problem is that splice site consensus sequences alone do not contain sufficient information to uniquely define splice junctions. Splice donor strength is often estimated by the degree to which it matches a 9nt consensus sequence at exon-intron boundaries. This is an imperfect measure because the consensus is sufficiently degenerate that most genes, in addition to their authentic splice sites, possess pseudo splice sites that match the consensus equally well but are nonfunctional. Indeed, both the authentic and cryptic donor sites in the α-globin gene have approximately equal strength by this criterion.
The solution to this problem involves additional regulatory sequences that enhance or inhibit the use of nearby consensus splice site motifs. Splicing enhancers and silencers are often studied in the context of alternative exons whose splicing must be regulated in the appropriate spatio-temporal patterns during development. However, it is important to remember that RNA binding proteins are ubiquitous along pre-mRNA sequences, where they play additional roles including suppression of pseudo splice sites. Even constitutively spliced exons may require splicing enhancer elements, an early example of which was described in human β-globin transcripts.6 These and other studies showed that protein coding sequences and splicing regulatory elements need to be coselected during evolution. Ji et al add to this list by finding that constitutive exons may also require enhancing elements in the flanking intron, such as the C-rich sequence, to promote efficient and accurate splicing.
Finally, the issue of splice site competition is an enormous challenge, not only for the splicing machinery but also for the scientist attempting to interpret the splicing consequences of single nucleotide variants in patients with genetic disease. Despite the wealth of exome and whole-genome sequencing data available from many patients, predicting which variants have deleterious effects on splicing is not trivial. About 9% of mutations in the Human Gene Mutation Database reportedly affect splicing,7 but this is almost certainly an underestimate because nonsense and missense mutations (and even translationally silent mutations) in the exons sometimes disrupt splicing.8 Moreover, mutations in introns can disrupt splicing patterns by creating new consensus splicing motifs or activating existing sites by improving the sequence context of existing cryptic splice sites. An example of the latter has been reported in patients with hereditary spherocytosis with aberrantly spliced α-spectrin pre-mRNA splicing, which is caused by an intronic mutation that strengthens a cryptic branchpoint motif so as to activate a nearby cryptic splice site.9 Recent approaches that use neural networks to predict splice sites on the basis of primary sequence alone may improve our ability to identify which among many sequence variants in a patient are most likely to cause disease.10
Conflict-of-interest disclosure: The author declares no competing financial interests.
