

Key Points
VWF heparin-binding domain binds to GFs.
VWF heparin-binding domain enhances angiogenesis in wound healing in collaboration with GFs.

Abstract
During wound healing, the distribution, availability, and signaling of growth factors (GFs) are orchestrated by their binding to extracellular matrix components in the wound microenvironment. Extracellular matrix proteins have been shown to modulate angiogenesis and promote wound healing through GF binding. The hemostatic protein von Willebrand factor (VWF) released by endothelial cells (ECs) in plasma and in the subendothelial matrix has been shown to regulate angiogenesis; this function is relevant to patients in whom VWF deficiency or dysfunction is associated with vascular malformations. Here, we show that VWF deficiency in mice causes delayed wound healing accompanied by decreased angiogenesis and decreased amounts of angiogenic GFs in the wound. We show that in vitro VWF binds to several GFs, including vascular endothelial growth factor-A (VEGF-A) isoforms and platelet-derived growth factor-BB (PDGF-BB), mainly through the heparin-binding domain (HBD) within the VWF A1 domain. VWF also binds to VEGF-A and fibroblast growth factor-2 (FGF-2) in human plasma and colocalizes with VEGF-A in ECs. Incorporation of the VWF A1 HBD into fibrin matrices enables sequestration and slow release of incorporated GFs. In vivo, VWF A1 HBD-functionalized fibrin matrices increased angiogenesis and GF retention in VWF-deficient mice. Treatment of chronic skin wounds in diabetic mice with VEGF-A165 and PDGF-BB incorporated within VWF A1 HBD-functionalized fibrin matrices accelerated wound healing, with increased angiogenesis and smooth muscle cell proliferation. Therefore, the VWF A1 HBD can function as a GF reservoir, leading to effective angiogenesis and tissue regeneration.
Introduction
von Willebrand factor (VWF) is a large plasma glycoprotein synthesized by endothelial cells (ECs) and megakaryocytes. It is best known for its role in hemostasis where it mediates platelet adhesion to the subendothelium at sites of endothelial damage and acts as a carrier to coagulation factor VIII (FVIII).1 Other functions reported for VWF include regulation of inflammation and angiogenesis.2 von Willebrand disease (VWD), the most common inherited bleeding disorder, is caused by defects or deficiency of VWF.3 In a subset of patients, vascular malformations in the gastrointestinal tract (ie, angiodysplasia) have been identified that can cause severe, intractable bleeding.2 Blood vessel formation and angiogenesis are crucial steps in wound healing. Delayed wound healing has been reported in some VWD patients,4 but whether VWF plays a role in wound healing remains unclear.
VWF comprises subunits made up of conserved modular domains in the following order: D1-D2-D′-D3-A1-A2-A3-D4-C1-C2-C3-C4-C5-C6-CK. Mature VWF is formed after multimerization and proteolytic release of the VWF propeptide5 (ie, the D1 and D2 domains). The A1 domain contains binding sites for platelet glycoprotein Ibα (GPIbα), heparin, and type I and type III collagen.6,7 The heparin-binding domain (HBD) within the A1 domain (Tyr1328-Ala1350 in the human sequence) shares the GPIbα-binding site and plays a crucial role in the regulation of platelet GPIbα binding.8,9
HBDs exist in a variety of extracellular matrix (ECM) proteins such as vitronectin, tenacin C, fibronectin, osteopontin, and fibrinogen.10-15 The HBDs of these proteins directly interact with heparin-binding growth factors (GFs),10-12 allowing the ECM to function as a GF reservoir.16,17 Binding between GFs and ECM proteins such as fibronectin controls local GF concentration, diffusion, and signaling.16,18 The control of GF activity and localization by ECM proteins is a key requirement for effective angiogenesis; indeed, a tissue gradient of vascular endothelial growth factor-A (VEGF-A) is the leading driver of angiogenic sprouting.19 ECM protein binding to GFs contributes to wound healing.16,17 Previously, we have shown that incorporation of the HBD and the integrin-binding domain of fibronectin (FN III9-10/12-14) or incorporation of the HBD from laminin within fibrin matrices improves the wound healing capacities of fibrin by promoting angiogenesis in the presence of VEGF-A165 and platelet-derived growth factor-BB (PDGF-BB).18,20
In this study, we hypothesize that VWF binds to heparin-binding GFs via its HBD, acting as a tissue reservoir for GFs and thus promoting effective angiogenesis and wound healing. We demonstrate broad VWF-GF binding and explore this in the molecular context of binding to VEGF-A and PDGF-BB in a model of dermal wound healing.
Materials and methods
Details regarding materials and methods are provided in the supplementary Data available online on the Blood Web site.
Results
VWF deficiency results in delayed wound healing and decreased angiogenesis
We tested whether endogenous VWF plays a role in dermal wound healing. Full-thickness back-skin wounds were made on VWF-deficient mice and littermate wild-type (WT) controls. After 5 days, wounds were analyzed (Figure 1). VWF deficiency significantly delayed wound closure; this was associated with poor granulation tissue formation (Figure 1A-C). VWF deficiency also decreased the proliferation of ECs and smooth muscle cells (SMCs) in the wounds, suggesting impaired angiogenesis (Figure 1D-E). At day 5 after wounding, VWF deficiency did not significantly affect the number of macrophages and neutrophils in the wounds (supplemental Figure 1A-B). The number of platelets was decreased in the VWF-deficient wounds, as expected (supplemental Figure 1C-E). We next measured the amount of VEGF-A and fibroblast growth factor-2 (FGF-2), both strong angiogenesis inducers,21 in the wounds. VWF deficiency decreased the amount of VEGF-A and FGF-2 in the wounds (Figure 1F-G). Immunostaining of wound tissue showed a diffuse distribution of VEGF-A in control mice, with enrichment around the blood vessels as expected; both were significantly decreased in wound tissues from VWF-deficient mice (Figure 1H-J; supplemental Figure 2). Interestingly, the concentration of VEGF-A in the plasma of VWF-deficient mice was increased and the concentration of FGF-2 showed a trend toward increase 5 days after wounding, suggesting that decreased amounts of GFs in the wound are not the result of a difference in circulating levels of VEGF-A and FGF-2 (supplemental Figure 3). These results suggest that VWF contributes to skin tissue repair through angiogenesis and GF involvement.
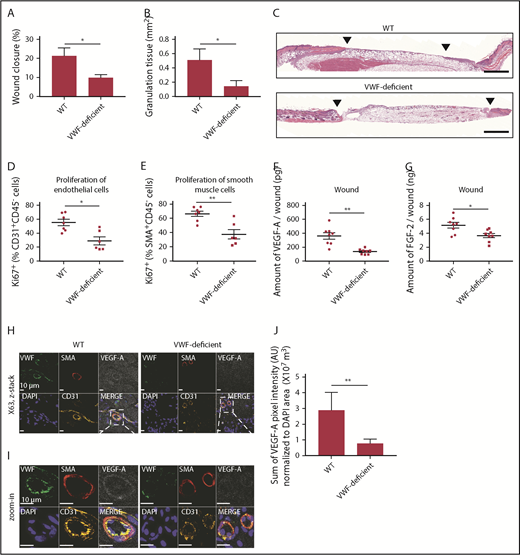
VWF-deficient mouse shows impaired wound healing and reduced angiogenesis. Full-thickness back-skin wounds were made in WT and VWF-deficient mice. After 5 days, (A) wound closure and (B) granulation tissue area were evaluated by histomorphometry. (WT, n = 6; VWF-deficient, n = 7). (C) Wound histology (hematoxylin and eosin staining). Black arrows indicate tips of the epithelium tongue. The granulation tissue (pink-violet) is characterized by infiltration of granulocytes with nuclei stained in dark-violet or black. Muscle under the wounds is stained in red; fat tissue appears as transparent bubbles. Scale bar = 800 µm. Proliferation of (D) CD31+CD45– ECs and (E) SMA+CD45– SMCs assessed by Ki67+ marker was determined using flow cytometry. The amounts of (F) VEGF-A and (G) FGF-2 in the wounds were quantified by enzyme-linked immunosorbent assay (ELISA). (H-I) Representative high-magnification image by immunofluorescence and (J) quantification of VEGF-A in mouse skin wound healing sections from WT and VWF-deficient mice; sections are costained for VWF (green), SMA (red), and CD31 (yellow) to visualize blood vessels; nuclei are identified by 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar = 10 μm. (J) Quantification represents the ratio between the sum of pixel intensity for VEGF-A signal and DAPI volume (μm3) (n = 2 fields per mouse; n = 5 mice per genotype). Graphical data are mean ± standard error of the mean (SEM), Statistical comparisons were carried out using the Mann-Whitney U test. AU, arbitrary unit. *P < .05; **P < .01.
VWF-deficient mouse shows impaired wound healing and reduced angiogenesis. Full-thickness back-skin wounds were made in WT and VWF-deficient mice. After 5 days, (A) wound closure and (B) granulation tissue area were evaluated by histomorphometry. (WT, n = 6; VWF-deficient, n = 7). (C) Wound histology (hematoxylin and eosin staining). Black arrows indicate tips of the epithelium tongue. The granulation tissue (pink-violet) is characterized by infiltration of granulocytes with nuclei stained in dark-violet or black. Muscle under the wounds is stained in red; fat tissue appears as transparent bubbles. Scale bar = 800 µm. Proliferation of (D) CD31+CD45– ECs and (E) SMA+CD45– SMCs assessed by Ki67+ marker was determined using flow cytometry. The amounts of (F) VEGF-A and (G) FGF-2 in the wounds were quantified by enzyme-linked immunosorbent assay (ELISA). (H-I) Representative high-magnification image by immunofluorescence and (J) quantification of VEGF-A in mouse skin wound healing sections from WT and VWF-deficient mice; sections are costained for VWF (green), SMA (red), and CD31 (yellow) to visualize blood vessels; nuclei are identified by 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar = 10 μm. (J) Quantification represents the ratio between the sum of pixel intensity for VEGF-A signal and DAPI volume (μm3) (n = 2 fields per mouse; n = 5 mice per genotype). Graphical data are mean ± standard error of the mean (SEM), Statistical comparisons were carried out using the Mann-Whitney U test. AU, arbitrary unit. *P < .05; **P < .01.
VWF binds to multiple GFs
Next, we tested the hypothesis that plasma-derived VWF protein binds to recombinant GF proteins. A panel of GFs from PDGF/VEGF, FGF, transforming growth factor-β (TGF-β)/bone morphogenetic protein (BMP), neurotrophin, and chemokine families were selected. The results of the binding screening are shown in Figure 2A-B. VWF bound to VEGF-A165, placenta growth factor 2 (PlGF-2), PDGF-AA, PDGF-BB, PDGF-CC, and PDGF-DD, but not to VEGF-A121 or PlGF-1, neither of which bind heparin. From the FGF family, VWF bound to FGF-2, FGF-7, and FGF-18, but not to FGF-1 or FGF-6. Among the TGF-β/BMP family, VWF showed strong binding to TGF-β1 and BMP-2, but not to TGF-β3 or BMP-7. Regarding the neurotrophins, both nerve growth factor-β (NGF-β) and neurotrophin-3 (NT-3) showed relevant binding. Neither insulin-like growth factor-I (IGF-I) nor IGF-II bound to VWF. In addition, epidermal growth factor (EGF) did not show binding to VWF. From the chemokine family, CXCL-11 and CXCL-12α did not bind strongly to VWF, whereas isoform CXCL-12γ, which has an additional HBD in its C-terminus,22,23 showed strong binding signal to VWF.22,23 VEGF-A121, which did not show significant binding to VWF by surface plasmon resonance (SPR) (supplemental Figure 4), was used as a nonbinding reference. These data indicate that VWF binds to multiple heparin binding GFs.
![Figure 2. Human plasma-derived VWF binds promiscuously to GFs with high affinity. VWF binding to (A) GFs and (B) chemokines were measured by ELISA. A450 nm represents absorbance at 450 nm. Signals from VEGF-A121 served as a baseline, and bovine serum albumin (BSA) served as a negative control (n = 4; data are mean ± standard deviation [SD]). Affinity (KD values are shown) of VWF against (C) VEGF-A165, (D) PDGF-BB, (E) NT-3, and (F) PDGF-DD was measured by surface plasmon resonance (SPR). SPR chips were functionalized with VWF (∼2000 resonance units [RU]), and the individual GF was flowed over the chips at indicated concentrations. Curves represent the specific responses (in RU) to VWF obtained. Experimental curves were fitted with (C,F) 1:1 Langmuir fit model and (D-E) heterogeneous ligand-parallel reactions binding. Binding kinetics values (dissociation constants [KD] and rate constants [kon and koff]) determined from the fitted curves are shown.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/24/10.1182_blood.2019000510/6/m_bloodbld2019000510f2.png?Expires=1769083867&Signature=bVx69dAUci3DAha9vC63Lwjo5M76kowJiaVRk9fZalrSRSm-fzqwRK89JEojibPba5kr8XgC8dZuu7sPilE0CGzbhgLKHC5Ahl9gSDpyNUhJOkU3FWpnP49ESGCX~dp0iS3d1uG0JZnADSQAaIe~OcNvifCPV6PtVJ-lWbHj8dAceYBA~R6JtQ3WHDdAqOZlCEctjwV-3DFxkarf1hTHgD6USoT8BssrhpkcvdmPlZ2HkyIx7AZU7HmDIotsD0q9TTjlcb8Yl8809d5dsieShac5dSLkoVv3p9BNtT-oS~~rAp3J4rkFYUqmRqVWHYW4QMLJLXULtqfx7bMUaIMINg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Human plasma-derived VWF binds promiscuously to GFs with high affinity. VWF binding to (A) GFs and (B) chemokines were measured by ELISA. A450 nm represents absorbance at 450 nm. Signals from VEGF-A121 served as a baseline, and bovine serum albumin (BSA) served as a negative control (n = 4; data are mean ± standard deviation [SD]). Affinity (KD values are shown) of VWF against (C) VEGF-A165, (D) PDGF-BB, (E) NT-3, and (F) PDGF-DD was measured by surface plasmon resonance (SPR). SPR chips were functionalized with VWF (∼2000 resonance units [RU]), and the individual GF was flowed over the chips at indicated concentrations. Curves represent the specific responses (in RU) to VWF obtained. Experimental curves were fitted with (C,F) 1:1 Langmuir fit model and (D-E) heterogeneous ligand-parallel reactions binding. Binding kinetics values (dissociation constants [KD] and rate constants [kon and koff]) determined from the fitted curves are shown.
Human plasma-derived VWF binds promiscuously to GFs with high affinity. VWF binding to (A) GFs and (B) chemokines were measured by ELISA. A450 nm represents absorbance at 450 nm. Signals from VEGF-A121 served as a baseline, and bovine serum albumin (BSA) served as a negative control (n = 4; data are mean ± standard deviation [SD]). Affinity (KD values are shown) of VWF against (C) VEGF-A165, (D) PDGF-BB, (E) NT-3, and (F) PDGF-DD was measured by surface plasmon resonance (SPR). SPR chips were functionalized with VWF (∼2000 resonance units [RU]), and the individual GF was flowed over the chips at indicated concentrations. Curves represent the specific responses (in RU) to VWF obtained. Experimental curves were fitted with (C,F) 1:1 Langmuir fit model and (D-E) heterogeneous ligand-parallel reactions binding. Binding kinetics values (dissociation constants [KD] and rate constants [kon and koff]) determined from the fitted curves are shown.
The binding affinity of VWF to GFs was determined by SPR (Figure 2C-F). The curves obtained for the specific binding signals were fitted with Langmuir binding kinetics. The binding affinity between VEGF-A165 and VWF was described by a single dissociation constant (KD value) of 27 nM. PDGF-BB had two estimated binding sites, with the lowest KD value of 24 nM. NT-3 also had two estimated binding sites, with the lowest KD value of 0.2 nM. PDGF-DD and VWF was described by a KD value of 13 nM. The nanomolar (nM) range of KD values demonstrates strong binding affinities of VWF to the heparin-binding GFs tested.
VWF colocalizes with VEGF-A in ECs and binds to GFs in human plasma
VEGF-A is produced, albeit at low levels, by ECs.24 We therefore investigated whether VEGF-A interaction with VWF resulted in colocalization within Weibel-Palade bodies (WPBs), endothelial storage organelles the assembly of which is dependent on VWF.25 In human umbilical vein ECs (HUVECs), VEGF-A was found to colocalize with VWF in WPB (Figure 3A-B). Stimulation of HUVECs with phorbol myristate acetate, which is known to release WPB content, resulted in release of both VWF and VEGF-A in the supernatant (Figure 3C-D). We tested the presence of the GF-VWF complex in plasma from a healthy donor. Immunoprecipitation followed by western blotting showed that VWF binds to VEGF-A and FGF-2 in human plasma (Figure 3E; supplemental Figure 5). These data suggest that GFs bind to VWF under physiological conditions.

VWF and GF colocalize in ECs and interact in human plasma. (A) Representative high-magnification immunofluorescence images of VWF (green) and VEGF-A (red) expression in the wound, shown as single grayscale channels, in HUVECs; nuclei are identified by DAPI (blue). The white box identifies zoom area shown in (B). Scale bar = 20 μm. (C) VWF was quantified by ELISA in total HUVEC lysates (left) or in cell culture supernatants (right) in the presence or absence of phorbol myristate acetate (PMA) stimulation. Data expressed as ng/mg are normalized to total protein levels (mg) (n = 4; data are mean ± SEM). (D) VEGF-A was quantified by ELISA in total HUVEC lysates (left) or in cell culture supernatants (right) in the presence or absence of PMA stimulation. Data expressed as pg/mg are normalized to total protein levels (mg) (n = 4; data are mean ± SEM). (E) Human plasma or positive control (recombinant VWF + recombinant VEGF-A or FGF-2) was subjected to immunoprecipitation with anti-human VEGF-A antibody or anti-human FGF-2 antibody. Western blotting was performed with collected proteins using anti-human VWF antibody. Mann-Whitney U test was used for analysis. *P < .05.
VWF and GF colocalize in ECs and interact in human plasma. (A) Representative high-magnification immunofluorescence images of VWF (green) and VEGF-A (red) expression in the wound, shown as single grayscale channels, in HUVECs; nuclei are identified by DAPI (blue). The white box identifies zoom area shown in (B). Scale bar = 20 μm. (C) VWF was quantified by ELISA in total HUVEC lysates (left) or in cell culture supernatants (right) in the presence or absence of phorbol myristate acetate (PMA) stimulation. Data expressed as ng/mg are normalized to total protein levels (mg) (n = 4; data are mean ± SEM). (D) VEGF-A was quantified by ELISA in total HUVEC lysates (left) or in cell culture supernatants (right) in the presence or absence of PMA stimulation. Data expressed as pg/mg are normalized to total protein levels (mg) (n = 4; data are mean ± SEM). (E) Human plasma or positive control (recombinant VWF + recombinant VEGF-A or FGF-2) was subjected to immunoprecipitation with anti-human VEGF-A antibody or anti-human FGF-2 antibody. Western blotting was performed with collected proteins using anti-human VWF antibody. Mann-Whitney U test was used for analysis. *P < .05.
HBD of VWF A1 domain binds to multiple GFs
We next investigated the domain within VWF responsible for association with GFs. Enzyme-linked immunosorbent assays (ELISAs) for VWF binding to VEGF-A165, PlGF-2, or FGF-2 were carried out in the presence of excess (10 µM) heparin. Excess heparin inhibited VWF binding to GFs (supplemental Figure 6), indicating involvement of HBDs. The HBD of VWF is located in the A1 domain (Figure 4A). Thus, we evaluated GF binding to recombinant A1 domain.9,26 VEGF-A165, PlGF-2, PDGF-BB, FGF-2, and CXCL-12γ showed strong binding to recombinant A1 domain, as measured by ELISA (Figure 4B). We next used a chemically synthesized VWF HBD (supplemental Table 1), the sequence of which was based on previous studies of the human VWF A1 domain.9,26 Importantly, this peptide reportedly does not bind to platelets.9 In these studies, VEGF-A165, PlGF-2, PDGF-BB, FGF-2, and CXCL-12γ showed binding to the VWF HBD, whereas neither VEGF-A121 nor PlGF-1 were able to bind to the VWF HBD (Figure 4C), consistent with the results for full-length VWF (Figure 2). These data show that the HBD of VWF A1 domain mediates VWF binding to GFs.
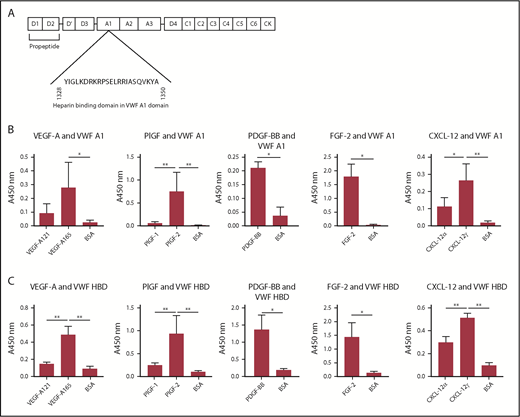
The HBD within the A1 domain of VWF mediates GF binding. (A) The location of the A1 domain and HBD within VWF. (B-C) Affinity of VEGF-A, PlGF, PDGF-BB, FGF-2, or CXCL-12 against (B) recombinant VWF A1 domain protein or (C) VWF A1 HBD peptide. ELISA plates were coated with 10 µg/mL recombinant VWF A1 domain protein or 10 µg/mL VWF A1 HBD peptide and further incubated with a 1-μg/mL VEGF-A, PlGF, PDGF-BB, FGF-2, or CXCL-12 solution. Bound GF or chemokine was detected by using a specific antibody for each GF or chemokine (n = 4; data are mean ± SD). Analysis of variance (ANOVA) with Tukey’s test or Mann-Whitney U test was performed. *P < .05; **P < .01.
The HBD within the A1 domain of VWF mediates GF binding. (A) The location of the A1 domain and HBD within VWF. (B-C) Affinity of VEGF-A, PlGF, PDGF-BB, FGF-2, or CXCL-12 against (B) recombinant VWF A1 domain protein or (C) VWF A1 HBD peptide. ELISA plates were coated with 10 µg/mL recombinant VWF A1 domain protein or 10 µg/mL VWF A1 HBD peptide and further incubated with a 1-μg/mL VEGF-A, PlGF, PDGF-BB, FGF-2, or CXCL-12 solution. Bound GF or chemokine was detected by using a specific antibody for each GF or chemokine (n = 4; data are mean ± SD). Analysis of variance (ANOVA) with Tukey’s test or Mann-Whitney U test was performed. *P < .05; **P < .01.
VWF binds to heparin-binding VEGF-A via the HBD within the A1 domain
Multiple isoforms of VEGF-A exist that differ in their binding to heparin and neuropilin-1 (supplemental Figure 7A). VEGF-A165, which contains an HBD, was found to bind plasma-derived purified VWF as well as immature, propeptide-containing recombinant VWF (supplemental Figure 7B). Similarly, VEGF-A145, which also contains a VEGF HBD, bound to VWF (supplemental Figure 7C), whereas VEGF-A121, which lacks an HBD, did not (supplemental Figure 7D). The VWF A1 domain bound to VEGF-A165 and VEGF-A145. However, no binding of the VWF A2 or A3 domains to VEGF-A165 or VEGF-A145 was detected (supplemental Figure 7B-C). The VWF A1 HBD peptide was also able to bind to VEGF-A165 and VEGF-A145 with a similar magnitude. Scrambling of the amino acid sequence of the VWF A1 HBD abolished the binding (supplemental Figure 7B-C), suggesting that the sequence, not just the total charge, is crucial for the association with VEGF-A165 and VEGF-A145. In addition, substitutions of Arg with Ser in the VWF A1 HBD sequence impaired the binding (supplemental Figure 7B-C), indicating that the positively charged residues are essential. These data demonstrate that the HBDs in the VWF A1 domain and in VEGF-A are responsible for binding between the 2 proteins.
VEGF-A does not affect VWF binding to platelet GPIb
The A1 domain of VWF contains the binding site for platelet GPIb, and this interaction mediates platelet recruitment to sites of injury.1 We therefore tested whether the association between VWF and VEGF-A may interfere with the binding between VWF and GPIbα by using a standard VWF ristocetin cofactor (VWF:RCo) assay.27 VEGF-A165, up to 10 µg/mL, did not affect VWF binding to platelet GPIb (supplemental Figure 8). These data suggest that the binding of VWF to VEGF-A165 does not alter the hemostatic function of VWF.
Type 2B VWD R1341 mutation impairs VWF binding to GF
Missense point mutations within the A1 domain of VWF have been reported in patients with type 2B VWD, a subtype in which the increased affinity of VWF for GPIbα results in spontaneous platelet aggregation, loss of the most active high-molecular-weight VWF multimers, thrombocytopenia, and bleeding.28-30 Type 2B mutations are clustered in exon 28 of the VWF gene, encoding the VWF A1 domain, and some map within the HBD.31 One such mutation that affects R1341 within the HBD has been reported in several type 2B VWD patients (von Willebrand factor Variant Database [VWFdb]; http://VWF.group.shef.ac.uk/), with substitutions to Leu, Pro, Gln, or Trp. Because Arg in HBDs seems to be crucial for GF binding (supplemental Figure 7B-C), we investigated whether this mutation could affect VWF-GF binding. Mutation of R1341 to any of these residues or to Ser abolished binding between the VWF A1 HBD and GFs (specifically, VEGF-A165, PDGF-BB, and FGF-2) (Figure 5A). These data indicate that the R1341 residue is indispensable for binding between VWF A1 HBD and GFs. Crucially, the R1341Q mutation also decreased binding of GFs (ie, VEGF-A165, PDGF-BB, and FGF-2) to full-length recombinant human VWF compared with WT (Figure 5B).
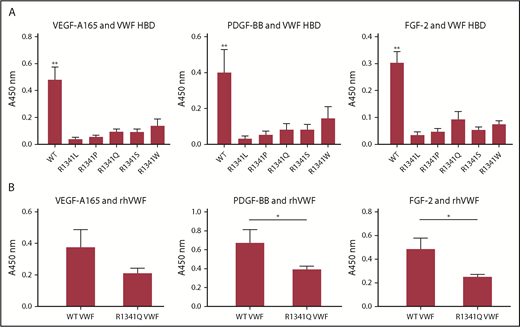
R1341 mutations observed in VWD type 2B patients impaires VWF-GF binding. (A) Binding of VEGF-A165, PDGF-BB, and FGF-2 to VWF A1 HBDs with R1341 substitutions. (n = 4; data are mean ± SD). (B) Binding of VEGF-A165, PDGF-BB, and FGF-2 to recombinant human VWF (rhVWF) with R1341Q substitution (n = 4; data are mean ± SD). ANOVA with (A) Tukey’s test and (B) Mann-Whitney U test. **P < .01; *P < .05.
R1341 mutations observed in VWD type 2B patients impaires VWF-GF binding. (A) Binding of VEGF-A165, PDGF-BB, and FGF-2 to VWF A1 HBDs with R1341 substitutions. (n = 4; data are mean ± SD). (B) Binding of VEGF-A165, PDGF-BB, and FGF-2 to recombinant human VWF (rhVWF) with R1341Q substitution (n = 4; data are mean ± SD). ANOVA with (A) Tukey’s test and (B) Mann-Whitney U test. **P < .01; *P < .05.
Retention of GFs in fibrin matrices is improved by incorporating VWF HBD peptide.
Next, we examined whether the VWF A1 HBD peptide can improve GF retention within a fibrin matrix, using VEGF-A165 and PDGF-BB, which are known to be quickly released from fibrin matrices.10,11,18 Fibrinogen solutions containing GFs and the VWF HBD with an integrated FXIIIa transglutaminase reactive substrate sequence18 (ie, α2PI1-8-VWF HBD) were polymerized to form a fibrin matrix using thrombin and FXIII. GF release from the matrix was determined by ELISA (Figure 6A-B). As previously shown, VEGF-A165 and PDGF-BB were quickly released from unmodified fibrin matrix (>85% released after 1 day).18 However, by incorporating the α2PI1-8-VWF HBD peptide, VEGF-A165 and PDGF-BB were retained within the fibrin matrices (45% and 52% retention on day 5, respectively). These results demonstrate that the VWF HBD enhances the function of a fibrin matrix as a GF reservoir and suggest that VWF HBD serves as a GF reservoir in multiple contexts and for multiple factors.
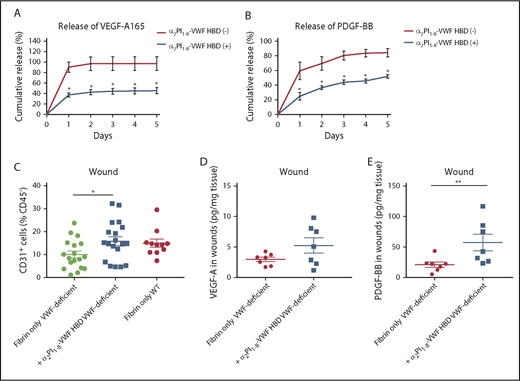
VWF HBD-functionalized fibrin matrices enhance GFs retention in vitro and in VWF-deficient mice. (A-B) GF retention in fibrin matrix. Graph showing the cumulative release of (A) VEGF-A165 or (B) PDGF-BB over 5 days (n = 4; data are mean ± SEM). (C-E) Full-thickness back-skin wounds in VWF-deficient mice were treated with fibrin only or fibrin functionalized with α2PI1–8-VWF HBD. (C) Five days after the wound treatment, the number of CD31+/CD45– ECs was determined by using flow cytometry (data are mean ± SEM). (D-E) The amounts of (D) VEGF-A and (E) PDGF-BB in the wounds were quantified by ELISA and normalized by the weight of the wound tissue (data are mean ± SEM). (A-B) Mann-Whitney U test for each time point, (C) ANOVA with Tukey’s test, and (D-E) Mann-Whitney U test. **P < .01; *P < .05.
VWF HBD-functionalized fibrin matrices enhance GFs retention in vitro and in VWF-deficient mice. (A-B) GF retention in fibrin matrix. Graph showing the cumulative release of (A) VEGF-A165 or (B) PDGF-BB over 5 days (n = 4; data are mean ± SEM). (C-E) Full-thickness back-skin wounds in VWF-deficient mice were treated with fibrin only or fibrin functionalized with α2PI1–8-VWF HBD. (C) Five days after the wound treatment, the number of CD31+/CD45– ECs was determined by using flow cytometry (data are mean ± SEM). (D-E) The amounts of (D) VEGF-A and (E) PDGF-BB in the wounds were quantified by ELISA and normalized by the weight of the wound tissue (data are mean ± SEM). (A-B) Mann-Whitney U test for each time point, (C) ANOVA with Tukey’s test, and (D-E) Mann-Whitney U test. **P < .01; *P < .05.
VWF HBD increased the density of ECs in VWF-deficient wounds
VWF deficiency in mice causes defective wound healing with reduced levels of GFs in the wound and reduced EC numbers (Figure 1). To test whether the VWF HBD was able to increase GF retention and enhance blood vessel formation in VWF-deficient mice, we applied fibrin matrices functionalized with or without VWF HBD in wounds of VWF-deficient mice. Five days after wounding, the levels of GFs and the number of CD31+ ECs in the wounds were analyzed. VWF HBD functionalized fibrin matrices significantly increased the number of CD31+ ECs to levels comparable to those of WT mice (Figure 6C). VEGF-A levels were increased by 1.8-fold compare with controls, although the difference was not statistically significant (Figure 6D). Levels of PDGF-BB in wound tissue were significantly increased in samples treated with the VWF HBD functionalized fibrin (Figure 6E). These data suggest that the VWF HBD promotes retention of endogenous GFs in the wound tissue and that this contributes to enhancing angiogenesis in VWF-deficient mice.
α2PI1-8-VWF HBD peptide functionalized fibrin matrix promotes chronic wound healing in vivo
Having established that the VWF HBD peptide is able to promote GF retention, we tested whether the peptide is able to potentiate the effect of GFs by inducing GF sequestration and slow release from matrices, thus enhancing skin wound healing in vivo. A genetic mouse model of type 2 diabetes provides a well-established and clinically relevant experimental system of delayed wound healing; induction of angiogenesis promotes wound healing in this model.32 VEGF-A165 and PDGF-BB, crucial angiogenesis inducers that exhibited binding to the VWF HBD, were incorporated within a fibrin matrix. As above, we used the FXIIIa-induced coupling of the α2PI1-8 sequence to fibrin with the α2PI1-8-VWF HBD to functionalize the matrix.18 Four groups of treatment were established: fibrin only, fibrin functionalized with α2PI1-8-VWF HBD, fibrin containing the GFs, and fibrin functionalized with α2PI1-8-VWF HBD containing the GFs. After 7 days, the histology of wounded skin was analyzed. The wounds that received fibrin matrices containing only GFs or VWF HBD did not differ from wounds treated with fibrin alone either in degree of wound closure or in amount of granulation tissue (Figure 7A-B). In contrast, the combined delivery of VEGF-A165 and PDGF-BB by fibrin functionalized with α2PI1-8-VWF HBD led to significantly faster wound closure as a result of re-epithelialization. The development of granulation tissue was maintained (Figure 7B). We next examined ECs in the wounds (Figure 7C). Codelivery of VEGF-A165 and PDGF-BB in fibrin functionalized with α2PI1-8-VWF HBD led to a significantly increased number of ECs compared with fibrin only after 5 days of wounding. Codelivery of VEGF-A165 and PDGF-BB in α2PI1-8-VWF HBD-functionalized fibrin significantly increased SMC proliferation (a marker of mature vessel formation) and were proliferated by PDGF-BB compared with fibrin only and α2PI1-8-VWF HBD-functionalized fibrin-only treatment groups (Figure 7D). These data show that treatment with α2PI1-8-VWF HBD and GFs incorporated within a fibrin matrix promotes wound closure by sequestration and slow release of VEGF-A165 and PDGF-BB.
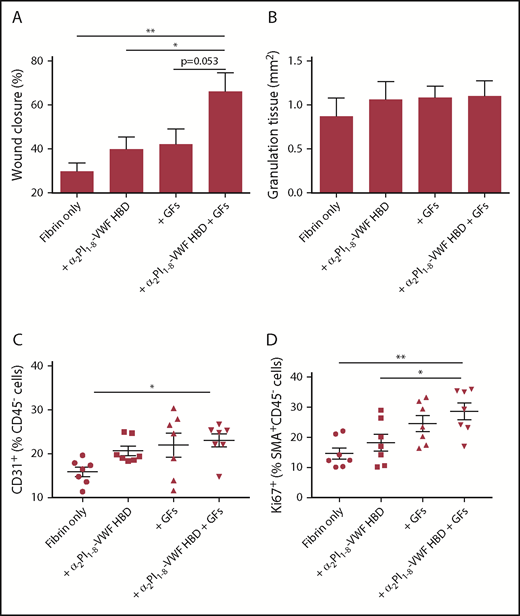
Delivering GFs within VWF HBD–functionalized fibrin matrices enhances skin wound healing in diabetic mice. Full-thickness back-skin wounds were treated with combined 100 ng of VEGF-A165 and 50 ng of PDGF-BB. Four groups were tested: fibrin only, fibrin functionalized with α2PI1–8-VWF HBD only, fibrin containing GFs only, and fibrin functionalized with α2PI1–8-VWF HBD containing GFs. After 7 days, (A) wound closure and (B) granulation tissue area were evaluated by histomorphometry (n = 11-13 per treatment group; data are mean ± SEM). Five days after the wound treatment, (C) the frequency of CD31+CD45– ECs within total alive cells and (D) proliferation of SMA+CD45– SMCs assessed by Ki67+ marker was determined by using flow cytometry (data are mean ± SEM). *P < .05; **P < .01, ANOVA with Tukey’s test.
Delivering GFs within VWF HBD–functionalized fibrin matrices enhances skin wound healing in diabetic mice. Full-thickness back-skin wounds were treated with combined 100 ng of VEGF-A165 and 50 ng of PDGF-BB. Four groups were tested: fibrin only, fibrin functionalized with α2PI1–8-VWF HBD only, fibrin containing GFs only, and fibrin functionalized with α2PI1–8-VWF HBD containing GFs. After 7 days, (A) wound closure and (B) granulation tissue area were evaluated by histomorphometry (n = 11-13 per treatment group; data are mean ± SEM). Five days after the wound treatment, (C) the frequency of CD31+CD45– ECs within total alive cells and (D) proliferation of SMA+CD45– SMCs assessed by Ki67+ marker was determined by using flow cytometry (data are mean ± SEM). *P < .05; **P < .01, ANOVA with Tukey’s test.
VWF HBD does not affect EC or fibroblast proliferation in vitro
To further investigate the mechanism through which the VWF HBD promotes wound healing, we tested its effect on fibroblast attachment and proliferation of fibroblasts and ECs. VWF HBD peptide coating modestly enhanced fibroblast attachment (supplemental Figure 9A); this effect was inhibited by adding 5 mM EDTA to the in vitro culture, suggesting that VWF HBD peptide may bind to cation-dependent cell adhesion receptors (supplemental Figure 9B). Coating of the VWF HBD peptide on cell culture plates did not significantly affect fibroblast proliferation in the presence of FGF-2, suggesting that the VWF HBD may slightly enhance cell adhesion, but not cell proliferation in vitro, at least in concert with this GF (supplemental Figure 9C). Similarly, VWF HBD did not affect EC proliferation in vitro (supplemental Figure 9D). These data indicate that, in the context of wound healing and tissue repair, the VWF HBD acts as a GF reservoir rather than as a cell scaffold, promoting effective wound healing and angiogenesis through its HBD.
Discussion
In this study, we show that VWF binds promiscuously to a variety of GFs via its HBD. We also show that a VWF HBD peptide is able to promote GF retention in vitro and in a wound healing model in vivo, suggesting that VWF acts as a reservoir for GFs, promoting wound healing partly through this mechanism.
Mapping of the VWF domain involved in GF binding identified the HBD in the A1 domain as the main GF binding site. However, mutation of R1341 in the A1 HBD domain did not completely abolish full-length VWF-GF binding, suggesting the possibility of other, minor GF binding domains. The best fit modeling in the SPR measurement of VWF-PDGF-BB and VWF-NT-3 was a heterogeneous ligand parallel reaction, also suggesting the existence of more than 1 GF-binding site in VWF. However, the ability of excess heparin to completely abrogate binding indicates that the main mechanism of VWF-GF association is via 1 or more HBDs. Further investigation will determine whether there are more GF binding domains (possibly other HBDs) within VWF.
Interestingly, most of the GFs that bind to VWF also bind fibrinogen, fibronectin, and laminin,11,18,20 suggesting a similar GF binding mechanism; however, VWF seems to bind some GFs more strongly than fibrinogen, because VEGF-A165 and PDGF-BB are quickly released from fibrin matrices.18 Only PDGF-CC showed specific binding to VWF, but not to fibrinogen or fibronectin. BMP-7, which binds specifically to fibronectin but not fibrinogen, did not show binding to VWF. In addition, VWF showed high affinities (KD in the nM range) toward GFs, suggesting that the binding capability of VWF is promiscuous yet high affinity.
VEGF-A isoforms reportedly have different levels of binding to the ECM21,33-35 and play different roles during vascular development and angiogenesis. For example, murine VEGF-A120 (121 in humans) does not bind to ECM proteins and alone is not sufficient to promote normal angiogenesis.34 Localized expression of VEGF-A is important in determining blood vessel branching architecture, because ectopic expression of VEGF-A in adult mice leads to the formation of disorganized networks, abnormal vessels, and convoluted, highly permeable structures.35-37 Such defects are the hallmark of vascular malformations; similar lesions (called angiodysplasia) are found in the gastrointestinal tract of patients with VWD and can cause significant bleeding that often does not respond to conventional treatments.38 The ability of VWF to bind GFs may be involved in the pathogenetic mechanism underlying angiodysplasia in VWD patients. We hypothesize that VWF is required for the physiological distribution and release of a GF gradient during blood vessel formation; in the absence of VWF, this gradient is disrupted leading to vascular malformations.
Previous studies have shown that VWF is required for effective angiogenesis; however, its role seems to be dependent on the model used and the microenvironment. VWF-deficient mice show enhanced constitutive vascularization in the ear and enhanced angiogenesis in Matrigel and in a model of ischemic stroke.39,40 However, in a hind limb ischemia model, VWF-deficient mice showed the opposite phenotype, namely reduced angiogenesis after ischemia.41 In the wound-healing model reported here, VWF deficiency was also associated with decreased angiogenesis. This apparent contradiction is not unique to VWF; several angiogenesis regulators (eg, the integrin αvβ3) have been shown to exert opposite effects in different models.42 In the case of VWF, the difference may be partly explained by the relationship between VWF and GFs. VWF-deficient ECs are more responsive to VEGF-A compared with control cells,40 and VEGF receptor 2 (VEGFR2) signaling is enhanced in ischemic brain microvasculature.39 However, in the wound healing model, VEGF-A levels are decreased in wounds of VWF-deficient mice, in line with decreased endothelial proliferation and angiogenesis.
Other factors may contribute to the delayed wound healing observed in VWF-deficient mice. Wound healing requires a complex coordination of coagulation, inflammation, and angiogenesis. Delayed wound healing was reported in FVIII-deficient mice.43 Although this may contribute to the phenotype in VWF-deficient mice, the initial coagulation response reportedly does not promote wound healing,44 and inhibition of VWF-platelet binding was reported not to delay wound healing.45 Moreover, no significant bleeding was observed in the majority of VWF mice when a wound was made on the upper back skin. Thus, although several mechanisms might contribute to the healing defect in VWF-deficient mice, our data indicate that the ability of VWF to bind and thus retain GFs in the wound is an important pathway through which VWF contributes to wound healing. Further studies will address in detail the possible contribution of other VWF-related pathways to this phenotype.
The VWF A1 HBD was able to enhance retention of GF in wounds in VWF-deficient mice and promote vascularization and SMC proliferation in the wounds of a type 2 diabetes model. These data indicate that VWF functions as a GF reservoir in the wounds and affects EC proliferation. Delayed wound healing has been reported in some VWD patients,4 but the incidence of the problem is unknown. Our findings suggest that a review of the clinical data related to wound healing in VWD patients would be timely; moreover, the ability of the VWF HBD to promote wound healing in the diabetic mouse model suggest exciting translational possibilities.
Our data show that VWF binds to GFs in plasma, suggesting that circulating VWF is able to serve as a carrier of multiple GFs. VWF is deposited at sites of tissue damage, interacting with subendothelial collagen to mediate platelet adhesion. Thus, VWF may contribute to local tissue repair and angiogenesis as well as hemostasis. The decrease in GFs in the VWF-deficient wounds could be the result of a combination of different reasons. In terms of the GF-producing cells, the number of ECs was reduced, but they do not produce much VEGF-A compared with VWF (Figure 3C-D). Thus, we suppose the contribution of ECs is limited. Other VEGF-A–producing cells such as macrophages are not significantly reduced in VWF-deficient wounds (supplemental Figure 1A). However, platelets are significantly reduced (supplemental Figure 1C-E), and they could be a source of VEGF-A. Thus, both the lack of VWF-dependent retention and of VEGF-A– and GF-producing cells could contribute to the decrease of GFs in VWF-deficient wounds.
GFs are considered to be crucial molecules for treating chronic diabetic ulcers. However, GFs have had only a modest impact on clinical practice to date.46,47 For example, VEGF-A, a crucial angiogenesis inducer, has not been approved by the US Food and Drug Administration because of ineffectiveness and adverse effects in clinical trials. PDGF-BB in Regranex has shown clinical efficacy, but there are still safety concerns such as cancer risk because of the high dose requirement.48 Therefore, engineering GF delivery systems for concentrating GF to wound sites and reducing GF doses could be a breakthrough for this issue. We show here that the effect of VEGF-A165 and PDGF-BB on tissue healing is significantly enhanced in combination with the covalently linked GF-binding domain derived from VWF via GF sustained retention, which suggests that the VWF HBD may be useful for therapeutic applications. A main advantage of using the VWF HBD peptide for this purpose, compared with the previous GF-binding domains from fibrinogen and fibronectin,11,18 is its ease of production: the HBD peptide can be chemically synthesized on a large scale with nonbiological origin and lower cost compared with recombinant proteins.
In conclusion, this study shows that VWF binds several GFs through the A1 HBD both in vitro and in human plasma. VWF deficiency in mice resulted in decreased angiogenesis and impaired wound healing, together with decreased amounts of VEGF-A and FGF-2 in the wound. Treatment of wounds in the VWF-deficient mice with fibrin matrices functionalized with VWF A1 HBD increased the amounts of GFs and promoted angiogenesis in the wound. Finally, in a diabetic mouse model, treatment of wounds by fibrin matrices functionalized with VWF A1 HBD containing low doses of GFs promoted angiogenesis and wound healing. These findings identify that VWF functions as a novel GF reservoir and promotes wound healing and also identify the VWF HBD as a possible novel tool to promote wound healing.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Human Tissue Resource Center at the University of Chicago for histology analysis and S. Gomes and P. S. Briquez for experimental assistance.
This work was supported in part by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant DP3DK108215 (J.A.H.) and a British Heart Foundation Intermediate Research Fellowship FS/10/47/28393 (R.D.S.).
Authorship
Contribution: J.I., R.D.S., A.M.R., and J.A.H. designed the project; J.I. and A.I. performed the experiments; J.I., A.I., R.D.S., T.A.J.M., M.A.L., A.M.R., and J.A.H. analyzed the data; J.I., A.M.R., and J.A.H. wrote the paper; K.S. and M.J.V.W. assisted with the wound-healing experiments; Y.T., K.F., and M.P.L. assisted with the GF retention experiments; R.D.S., T.A.J.M., K.E.S., and M.A.L. assisted with providing VWF samples and VWF-deficient mice; C.R.P. performed the VWF-VEGF colocalization study; and T.A.J.M. performed the platelet binding study.
Conflict-of-interest disclosure: The authors’ institutions (University of Chicago and Imperial College London) filed for patent protection on aspects of the VWF HBD and its uses, and J.I., A.I., J.A.H., R.D.S., and A.M.R. are named as inventors on that patent application. The remaining authors declare no competing financial interests.
Correspondence: Anna M. Randi, Vascular Sciences, National Heart and Lung Institute, Hammersmith Campus, Imperial College London, London W12 ONN, United Kingdom; e-mail: a.randi@imperial.ac.uk; and Jeffrey A. Hubbell, 5640 S. Ellis Ave, Institute for Molecular Engineering, University of Chicago, Chicago, IL 60637; e-mail: jhubbell@uchicago.edu.
