

Key Points
Engineered Bcor truncation mutations collaborate with NP23 fusion to generate pro-B1 ALL.
NP23/Bcor pro-B1 ALL acquire somatic Jak mutations and are sensitive to therapy with Jak inhibitors in vitro and in vivo.

Abstract
Approximately 10% of NUP98-PHF23 (NP23) mice develop an aggressive acute lymphoblastic leukemia of B-1 lymphocyte progenitor origin (pro-B1 ALL), accompanied by somatic frameshift mutations of the BCL6 interacting corepressor (Bcor) gene, most commonly within a 9-bp “hotspot” in Bcor exon 8. To determine whether experimentally engineered Bcor mutations would lead to pro-B1 ALL, we used clustered, regularly interspaced, short palindromic repeats–associated protein 9 to introduce a Bcor frameshift mutation into NP23 hematopoietic stem and progenitor cells through the use of Bcor small guide RNAs (Bcor sgRNAs). Recipient mice transplanted with NP23 bone marrow or fetal liver cells that had been transduced with a Bcor sgRNA developed pro-B1 ALL, characterized by a B-1 progenitor immunophenotype, clonal Igh gene rearrangement, and Bcor indel mutation, whereas control recipients did not. Similar to a subset of human B-cell precursor ALL, the murine pro-B1 ALL had acquired somatic mutations in Jak kinase genes. JAK inhibitors (ruxolitinib and tofacitinib) inhibited the growth of pro-B1 ALL cell lines established from Bcor sgRNA/NP23 recipients at clinically achievable concentrations (100 nM). Our results demonstrate that Bcor mutations collaborate with NP23 to induce pro-B1 ALL, and that JAK inhibitors are potential therapies for pro-B1 ALL.
Introduction
NUP98-PHF23 (NP23) transgenic mice develop progenitor B-1 acute lymphoblastic leukemia (pro-B1 ALL) with the immunophenotype (B220lo/−/CD19+/AA4.1+).1,2 Whole-exome sequencing showed that all pro-B1 ALL samples had acquired indel1 mutations of the Bcor gene, leading to the introduction of premature stop codons. Of note, most of these acquired mutations occurred within a 9-bp “hotspot” in Bcor exon 8, suggesting that these Bcor mutations may be important for leukemic transformation.1,2 Moreover, >70% of NP23 pro-B1 ALL acquired mutations in the Jak/Stat pathway.1 The NP23 murine pro-B1 ALLs are similar to human B-cell precursor (BCP) ALL with CRLF2 rearrangements in terms of Crlf2 expression, gene expression profile,1 VH-region usage,1 and acquired, complementary JAK mutations.3-6 Although BCOR mutations are rare in human BCP ALL, recurrent BCOR mutations, primarily SNV, indels, and gene fusions are found in a wide spectrum of human malignancy, including chronic lymphocytic leukemia7,8 and acute myeloid leukemia (AML).9
The clustered, regularly interspaced, short palindromic repeats (CRISPR)–associated protein (Cas) systems10 have been successfully used to introduce targeted loss-of-function mutations at specific sites in the genome in multiple model organisms11-16 ; mouse models of myeloid malignancy have used CRISPR-Cas9 to inactivate tumor suppressor genes.17 In this study, we use CRISPR-Cas9 to induce Bcor frameshift mutations in hematopoietic precursors leading to pro-B1 ALL.
Study design
Guide RNA plasmids and lentiviral particle production
Bcor small guide RNAs (sgRNAs) were cloned into the BsmbI site of pL-sgRNA-cas9-GFP vector, and particles generated by cotransfection with psPAX2 and pMD2.G into 293T cells.
Mice and transplantation
Lineage depleted fetal liver (FL) or bone marrow (BM) was transduced with empty vector (EV) or Bcor sgRNA lentiviral particles and transplanted into lethally irradiated (900 cGy) recipients. Recipients showing signs of leukemia were humanely euthanized. All animal experiments were approved by the National Cancer Institute Animal Care and Use Committee.
Jak inhibitor treatment
NP23/Bcor cell lines with acquired Jak mutations were treated with ruxolitinib or tofacitinib (Selleck Chemicals). Cell number was determined by trypan blue exclusion (see supplemental Materials and methods, available on the Blood Web site, for additional details).
Results and discussion
Use of CRISPR/cas9 to induce frameshift mutations at Bcor “hotspot”
To mimic the somatic frameshift mutation of Bcor that occurred in pro-B1 ALL, we designed Bcor sgRNAs close to the 9-bp Bcor “hotspot” (supplemental Figure 1A-B). sgRNAs were cloned into the pL-sgRNA-cas9-GFP vector and transduced into NIH3T3 cells. DNA was harvested and the relevant region of Bcor was amplified (supplemental Figure 1C). Sequencing chromatograms show multiple superimposed sequences, near the targeted PAM sequence (supplemental Figure 1D), reflecting sgRNA-induced indels (supplemental Figure 1E). To demonstrate that Bcor sgRNA could edit the genomes of primary mouse hematopoietic stem and progenitor cells (HSPCs) ex vivo, we transduced purified lineage negative (Lin−) BM or FL HSPCs. Bcor indel mutations were identified in both FL and BM HSPC transduced with Bcor sgRNA1 (supplemental Figure 1F).
Although the generation of Bcor indels may not be highly efficient, we reasoned that a transformed, leukemic clone would have a growth advantage and be selected in vivo. Lin− HSPC cells were isolated from WT and NP23 BM (age 1-3 months) or FL (E14.5 days), transduced with Bcor sgRNA1 or empty vector, and transplanted into lethally irradiated wild-type (WT) congenic recipients (supplemental Figure 2A). Mice were cotransplanted with a radiation-sparing dose of 2 × 10E05 WT BM cells that expressed CD45.1, which allowed distinction from the transduced BM or FL cells that express CD45.2. Serial analysis of peripheral blood demonstrated an expansion of CD45.2+ and CD19+B220lo/− cells over a period of 6 months in NP23/Bcor sgRNA1 recipients (supplemental Figure 2B-C).
NP23/Bcor recipients develop pro-B1 ALL
Six NP23/Bcor recipients and 1 NP23/EV recipient developed leukemia (Figure 1A) characterized by hunched posture, lethargy, and abnormal complete blood counts (Figure 1B; supplemental Table 1). Flow cytometry revealed infiltration of BM and spleen with leukemic cells that were CD19+B220lo/− (Figure 1C). B220 expression in the leukemic clone, although variable, was consistently lower than the B220 expression of residual normal splenic B cells (Figure 1C). BM cytospin and peripheral blood showed numerous lymphoblasts (Figure 1D). All NP23/Bcor leukemias showed indel mutations within 10 bp of the PAM site, as anticipated for CRISPR-induced indels (supplemental Figure 3A). Nucleotide sequence of subcloned polymerase chain reaction (PCR) products showed that overlapping sequences were due to contamination of indel alleles with WT sequence (#A1929) or 2 independent Bcor mutations (#A1942).
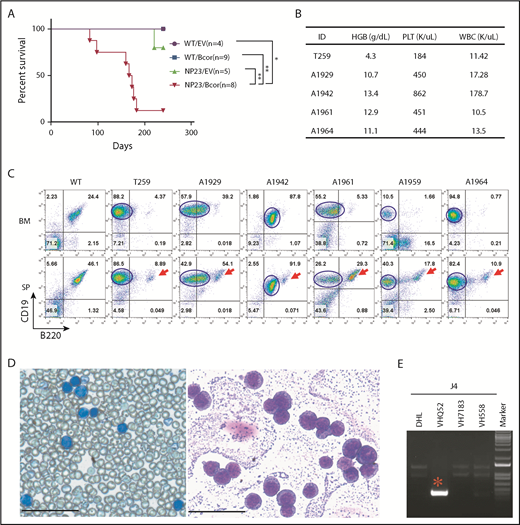
Introduction of Bcor frameshift mutations with CRISPR/Cas9 lead to pro-B1 ALL in NP23 HSPCs. (A) Survival curve for recipient mice transplanted with WT/EV, WT/Bcor sgRNA1 (Bcor), NP23/EV, and NP23/Bcor lin− BM or FL cells. Survival curve represents pooled results from 6 experiments and includes recipients of transduced cells (defined as GFP PCR+ and >1% CD45.2+ at 1 month posttransplantation). P value is indicated (log-rank Mantel-Cox test), *P < .05; **P < .01. (B) Complete blood count (CBC). (C) Flow cytometry of BM and spleen from mice WT, T259, A1929, A1942, A1961, A1959, and A1964 stained with B220, CD19, and CD45.2 antibodies. Red arrow indicates residual normal B cells; blue line outlines the malignant pro-B1 population. Expression of B220 and CD19 were analyzed by gating CD45.2+ live cells in the A1929, A1942, and A1961 mice, whereas the expression of B220 and CD19 were analyzed by gating live cells in the T259, A1959, and A1964 mice. (D) May-Grunwald Giemsa–stained peripheral blood (left) and BM cytospin (right) from mouse T259. Scale bars, 50 μm. (E) Igh DJ and VDJ rearrangement assay for the malignant cells of T259. *Clonal Igh rearrangement. HGB, hemoglobin; PLT, platelet; SP, spleen; WBC, white blood cell.
Introduction of Bcor frameshift mutations with CRISPR/Cas9 lead to pro-B1 ALL in NP23 HSPCs. (A) Survival curve for recipient mice transplanted with WT/EV, WT/Bcor sgRNA1 (Bcor), NP23/EV, and NP23/Bcor lin− BM or FL cells. Survival curve represents pooled results from 6 experiments and includes recipients of transduced cells (defined as GFP PCR+ and >1% CD45.2+ at 1 month posttransplantation). P value is indicated (log-rank Mantel-Cox test), *P < .05; **P < .01. (B) Complete blood count (CBC). (C) Flow cytometry of BM and spleen from mice WT, T259, A1929, A1942, A1961, A1959, and A1964 stained with B220, CD19, and CD45.2 antibodies. Red arrow indicates residual normal B cells; blue line outlines the malignant pro-B1 population. Expression of B220 and CD19 were analyzed by gating CD45.2+ live cells in the A1929, A1942, and A1961 mice, whereas the expression of B220 and CD19 were analyzed by gating live cells in the T259, A1959, and A1964 mice. (D) May-Grunwald Giemsa–stained peripheral blood (left) and BM cytospin (right) from mouse T259. Scale bars, 50 μm. (E) Igh DJ and VDJ rearrangement assay for the malignant cells of T259. *Clonal Igh rearrangement. HGB, hemoglobin; PLT, platelet; SP, spleen; WBC, white blood cell.
Pro-B1 ALLs showed clonal Igh gene rearrangements (Figure 1E; supplemental Figure 3B-D); similar to the previously reported pro-B1 ALL,1 the VH gene segment usage was biased toward the most 3′ VH segments, a known property of fetal liver B-1 lymphocytes.1,18 Samples A1942, A1961, A1959, and A1964 had 2 clonal Igh VDJ or DJ rearrangements (supplemental Figure 3B-C), consistent with subclonal heterogeneity of the tumor cell populations. In 5 of 6 samples, tissue sections showed a perivascular invasion of the liver or lung (supplemental Figure 4), and DNA extracted from the invaded tissue showed clonal Igh gene rearrangements identical to those found in leukemic cells from BM or peripheral blood.
Curiously, all leukemias were GFP− (supplemental Figure 5A), and some were CD45.2−. However, Cas9, translated from the same primary transcript as GFP, was also silenced (supplemental Figure 5B), despite the fact that PCR of genomic DNA documented the presence of both GFP and Cas9 DNA in the pro-B1 leukemias (supplemental Figure 5C) as well as the NP23 transgene (supplemental Figure 5D). Taken together, these results indicated that both GFP and CD45.2 could be downregulated in the leukemic cells, and that GFP fluorescence may not be a reliable marker for leukemic cells using this system.
Concurrent pre-T LBL and pro-B1 ALL or AML and pro-B1 ALL in recipient mice
Mouse #A1942 developed a precursor T-cell lymphoblastic leukemia/lymphoma (pre-T LBL), characterized by thymoma, CD4+CD8− expansion (supplemental Figure 6A), and clonal Tcrb rearrangement (supplemental Figure 6B), in addition to the pro-B1 ALL. Supplemental Figure 6C shows that the CD4+CD8− pre-T LBL arose from an unrelated clone because it had a Bcor indel different than the pro-B1 ALL. This finding is consistent with a report that indicates Bcor internal deletions predispose mice to pre-T LBL.19 Similarly, mouse #1959 developed a concurrent AML (supplemental Figure 6D) unrelated to the pro-B1 ALL; this AML was WT for Bcor (supplemental Figure 6E).
NP23/Bcor pro-B1 ALL acquire JAK mutations and are sensitive to JAK inhibitors
Whole-exome sequencing revealed spontaneous missense mutations of Jak2 or Jak3 in samples #T259 and #A1929 (supplemental Tables 2 and 3; supplemental Figure 7A-B). Immortal cell lines that retained the CD19+B220lo/−AA4.1+CD43+ immunophenotype characteristic of pro B1 lymphocytes were established from 3 pro-B1 ALL samples (Figure 2A-B; supplemental Figures 8A-B and 9), and transduction of these cell line with full length Bcor inhibited their growth (supplemental Figure 10). Given that childhood BCP ALL patients with CRLF2 rearrangements have features consistent with a pro-B1 origin,1 and that many of these patients have JAK mutations,4-6 we wished to determine if NP23/Bcor cell lines could be used to study pro-B1 ALL therapy. JAK inhibitors ruxolitinib20-22 or tofacitinib23,24 inhibited the growth of pro-B1 ALL cell lines in vitro (Figure 2C-E), and induced apoptosis at concentrations as low as 10 nM (supplemental Figure 11A-B). Treatment of the T259 cell line with tofacitinib in vivo showed decreased phospho-Stat5 (supplemental Figure 12A), a trend (P = .06) toward decreased numbers of circulating leukemic cells (supplemental Figure 12B-C) and increased survival (Figure 2F) in the treatment group. These results are similar to previously reported in vivo findings that suggested that monotherapy with Jak inhibitors was only partially effective at eliminating BCP ALL and that combination therapies would be more effective.25
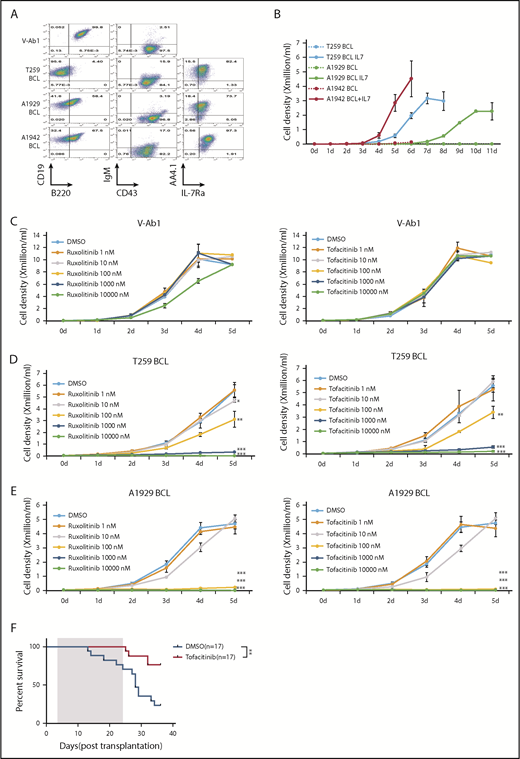
NP23/Bcor pro-B1 ALL cell lines are sensitive to JAK inhibitors. (A) V-Ab1, T259, A1929, and A1942 B-cell line (pro-B1 cell line) were stained with the indicated antibodies. Expression of AA4.1 and IL7-Ra were detected when B-cell line were cultured in RPMI medium without IL7 and serum for 6 hours. (B) Growth curve for T259, A1929, and A1942 B-cell line. A total of 1 × 104 T259, A1929, and A1942 B-cell lines were plated and cultured for 11 days in RPMI medium with or without 5 ng/mL IL-7. Growth curves of a v-Abl transformed B-cell line (C), T259 B-cell line (D), and A1929 B-cell line (E) treated with ruxolitinib (left) or tofacitinib (right) for 5 days. Cell number was determined by trypan blue exclusion. Each point represents the mean ± standard deviation (n = 3). Student t test: *P < .05; **P < .01; ***P < .001. (F) Survival curve for leukemic mice treated with tofacitinib. Mice were transplanted with 1 × 106 T259 B-cell line and treated daily for 21 days (treatment duration shaded in gray), beginning day 3 posttransplant, with either 40 mg/kg/d tofacitinib or vehicle control via oral gavage. P value is indicated (log-rank Mantel-Cox test). DMSO, dimethyl sulfoxide.
NP23/Bcor pro-B1 ALL cell lines are sensitive to JAK inhibitors. (A) V-Ab1, T259, A1929, and A1942 B-cell line (pro-B1 cell line) were stained with the indicated antibodies. Expression of AA4.1 and IL7-Ra were detected when B-cell line were cultured in RPMI medium without IL7 and serum for 6 hours. (B) Growth curve for T259, A1929, and A1942 B-cell line. A total of 1 × 104 T259, A1929, and A1942 B-cell lines were plated and cultured for 11 days in RPMI medium with or without 5 ng/mL IL-7. Growth curves of a v-Abl transformed B-cell line (C), T259 B-cell line (D), and A1929 B-cell line (E) treated with ruxolitinib (left) or tofacitinib (right) for 5 days. Cell number was determined by trypan blue exclusion. Each point represents the mean ± standard deviation (n = 3). Student t test: *P < .05; **P < .01; ***P < .001. (F) Survival curve for leukemic mice treated with tofacitinib. Mice were transplanted with 1 × 106 T259 B-cell line and treated daily for 21 days (treatment duration shaded in gray), beginning day 3 posttransplant, with either 40 mg/kg/d tofacitinib or vehicle control via oral gavage. P value is indicated (log-rank Mantel-Cox test). DMSO, dimethyl sulfoxide.
These findings demonstrate that CRISPR-induced Bcor frameshift collaborates with an NP23 transgene to predispose B-1 progenitors to leukemic transformation. These 2 events are unlikely to be sufficient for leukemic transformation because we detected spontaneous Jak pathway mutations that were required for continued growth of the leukemic cells. This constellation of mutations (NP23 expression leading to increased stem cell self-renewal, Bcor frameshift leading to impaired B-cell differentiation, and Jak pathway mutations leading to dysregulated proliferation) is similar to that seen in human BCP ALL patients,3 and suggests that the NP23/Bcor mutant mice and cell lines will be a useful model for human pro-B1 ALL.
Presented in abstract form at the 60th annual meeting of the American Society of Hematology, San Diego, CA, 1 December 2018.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Michael Kuehl, Avinash Bhandoola, and current and former members of Aplan laboratory for insightful discussions. They also thank the National Cancer Institute (NCI) Sequencing Minicore for Sanger sequencing, the NCI Transgenic Core for generation of transgenic mice, the NCI Flow Cytometry Core for cell sorting, Shelley Hoover and Mark Simpson of the NCI Molecular Pathology Unit for assistance with slide imaging, Vivian Bardwell and Charles Hemenway for kindly providing Bcor plasmids, and Maria Jorge for excellent animal husbandry.
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute (ZIA SC 010378 and BC 010983).
Authorship
Contribution: P.D.A. and M.Y. conceived and designed the project; M.Y., Y.J.C., and R.L.W. performed experiments; B.L.E. and R.C.L. contributed vital reagents and protocols; and M.Y., Y.J.C., R.L.W., P.S.M., Y.J.Z., and P.D.A. analyzed the data; M.Y. wrote the initial draft of the manuscript; and all authors reviewed drafts of the manuscript.
Conflict-of-interest disclosure: B.L.E. has received funding from Celgene and Deerfield and consulting fees from GRAIL. R.C.L. has received funding from MedImmune and Jazz and consulting fees from Takeda. The remaining authors declare no competing financial interests.
Correspondence: Peter D. Aplan, Genetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Building 37 Room 6002, 37 Convent Dr, Bethesda, MD 20892; e-mail: aplanp@mail.nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal