

Key Points
Dose finding established venetoclax 400 mg combined with obinutuzumab; this regimen had an acceptable safety profile in R/R and 1L CLL.
Venetoclax-obinutuzumab elicited high response rates with deep remissions in R/R and 1L CLL, irrespective of cytogenetic risk factors.

Abstract
This single-arm, open-label, phase 1b study evaluated the maximum tolerated dose (MTD) of venetoclax when given with obinutuzumab and its safety and tolerability in patients with relapsed/refractory (R/R) or previously untreated (first line [1L]) chronic lymphocytic leukemia (CLL). Venetoclax dose initially was escalated (100-400 mg) in a 3 + 3 design to define MTD combined with standard-dose obinutuzumab. Patients received venetoclax (schedule A) or obinutuzumab (schedule B) first to compare safety and determine dose/schedule for expansion. Venetoclax-obinutuzumab was administered for 6 cycles, followed by venetoclax monotherapy until disease progression (R/R) or fixed duration 1-year treatment (1L). Fifty R/R and 32 1L patients were enrolled. No dose-limiting toxicities were observed. Safety, including incidence of tumor lysis syndrome (TLS), did not differ between schedules (2 laboratory TLSs per schedule). Schedule B and a 400-mg dose of venetoclax were chosen for expansion. The most common grade 3-4 adverse event was neutropenia (R/R, 58% of patients; 1L, 53%). Rates of grade 3-4 infections were 29% (R/R) and 13% (1L); no fatal infections occurred in 1L. All infusion-related reactions were grade 1-2, except for 2 grade 3 events. No clinical TLS was observed. Overall best response rate was 95% in R/R (complete response [CR]/CR with incomplete marrow recovery [CRi], 37%) and 100% in 1L (CR/CRi, 78%) patients. Rate of undetectable (<10−4) minimal residual disease (uMRD) in peripheral blood for R/R and 1L patients, respectively, was 64% and 91% ≥3 months after last obinutuzumab dose. Venetoclax and obinutuzumab therapy had an acceptable safety profile and elicited durable responses and high rates of uMRD. This trial was registered at www.clinicaltrials.gov as #NCT01685892.
Introduction
Despite the evolving therapeutic landscape,1,2 chronic lymphocytic leukemia (CLL) remains incurable; most patients relapse or become treatment refractory.3-6 Novel targeted agents (B-cell receptor inhibitors) are used mainly in high-risk patients, especially where standard chemoimmunotherapy may be unsuitable due to toxicity and short remission durations. Although these novel agents improve progression-free survival (PFS), they often require prolonged treatment leading to unique toxicities.7-9 Further investigation of chemotherapy-free regimens, particularly with a fixed duration of treatment, is warranted in previously untreated (first line [1L]) and relapsed/refractory (R/R) CLL.
B-cell lymphoma 2 (BCL-2) overexpression allows CLL cells to evade apoptosis by sequestering proapoptotic proteins,10 thereby representing a therapeutic target. Venetoclax, a potent oral BCL-2 inhibitor,11 acts independently of TP5312 and has demonstrated substantial anti-CLL activity as monotherapy,13,14 and with rituximab,15,16 in R/R CLL patients. Fixed-duration venetoclax-rituximab improved PFS vs bendamustine-rituximab (BR) and achieved undetectable minimal residual disease (uMRD) in 62.4% of R/R CLL patients in the phase 3 MURANO study.16,17
Obinutuzumab, a type II anti-CD20 antibody,18 is also active in CLL.19-21 Preclinically, obinutuzumab mediates superior B-cell depletion vs rituximab in whole blood from CLL patients, independent of prognostic markers.18,22 Clinically, obinutuzumab-chlorambucil was associated with a PFS and overall survival benefit over rituximab-chlorambucil in the phase 3 CLL11 trial,20,23 leading to the approval of obinutuzumab-chlorambucil as a frontline therapy for CLL patients with comorbidities.24
Preclinical assessments to evaluate venetoclax-obinutuzumab as proof of concept showed that reduction of B cells was significantly higher with venetoclax-obinutuzumab than with venetoclax-rituximab or venetoclax alone (supplemental Table 1 and supplemental Figure 1, available on the Blood Web site). Here, we report results from a phase 1b study with venetoclax-obinutuzumab in R/R and 1L CLL (NCT01685892).
Patients and methods
Study conduct
This phase 1b, single-arm, open-label study was conducted at 11 sites across the United States and the United Kingdom. Review boards at all institutions approved the protocol. Patients provided written informed consent.
Patients
Eligible patients (supplemental Table 2) were aged ≥18 years with CLL in need of therapy by International Workshop on CLL (iwCLL) 2008 criteria25 and had: an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0-1; adequate hematologic function unless directly attributable to underlying CLL; and adequate organ function, including creatinine clearance ≥30 mL/min. Patients with R/R CLL must have received 1 to 3 prior chemotherapy-containing regimens; patients with 17p deletion (del[17p]) and/or TP53 mutation could have received at least 1 line of prior therapy with alemtuzumab-containing treatment or a B-cell receptor inhibitor (ibrutinib or idelalisib).
Study design and treatment
The study comprised 2 phases for each patient population (R/R and 1L): dose finding and safety expansion (supplemental Figure 2). Dose finding was planned to include multiple doses of venetoclax (100-600 mg) combined with standard-dose obinutuzumab (cycle 1: 100 mg day 1, 900 mg day 2, 1000 mg days 8 and 15; cycles 2-6: 1000 mg day 1) in 28-day cycles. Ultimately, the 600-mg dose was not explored after review of the present study and program-wide data, including data review of a phase 1b study in CLL with venetoclax-rituximab, in which the recommended phase 2 dose of venetoclax was 400 mg.15 To mitigate risk of tumor lysis syndrome (TLS), venetoclax was initiated with a ramp-up period with weekly dose increases to target dose (Figure 1). Prophylactic measures for TLS mitigation included hydration, allopurinol, rasburicase (for TLS high-risk patients with high pretreatment uric acid levels), and hospitalization for the first venetoclax dose (supplemental Table 3).
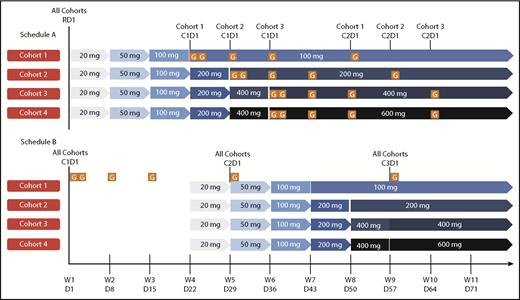
Dosing schedule. Schedule A, Venetoclax followed by obinutuzumab. Schedule B, Obinutuzumab followed by venetoclax. For both the R/R and 1L populations, schedule A was examined prior to schedule B. Data from schedule A provided safety guidance for subsequent dose finding for patients in schedule B after a data review by an internal monitoring safety team and a scientific overview committee. Venetoclax ramp-up: 3 weeks for the 100-mg cohort, 4 weeks for the 200-mg cohort, and 5 weeks for the 400-mg cohort; each cohort dose was continued for a total of 12 months with potential for extension if BM MRD+ or PR (1L) or until disease progression (R/R); venetoclax plus obinutuzumab (6 cycles), then venetoclax monotherapy. Cohort 4 (600 mg) was planned but not explored. Venetoclax ramp-up and maximum cohort dose are indicated by the blue arrows. Obinutuzumab dosing schedule: C1D1, 100 mg; C1D2, 900 mg; C1D8 and 15, 1000 mg; C2-6D1, 1000 mg. C, cycle; D, day; G, GA101/obinutuzumab; PR, partial response; RD1, ramp-up day 1; W, week.
Dosing schedule. Schedule A, Venetoclax followed by obinutuzumab. Schedule B, Obinutuzumab followed by venetoclax. For both the R/R and 1L populations, schedule A was examined prior to schedule B. Data from schedule A provided safety guidance for subsequent dose finding for patients in schedule B after a data review by an internal monitoring safety team and a scientific overview committee. Venetoclax ramp-up: 3 weeks for the 100-mg cohort, 4 weeks for the 200-mg cohort, and 5 weeks for the 400-mg cohort; each cohort dose was continued for a total of 12 months with potential for extension if BM MRD+ or PR (1L) or until disease progression (R/R); venetoclax plus obinutuzumab (6 cycles), then venetoclax monotherapy. Cohort 4 (600 mg) was planned but not explored. Venetoclax ramp-up and maximum cohort dose are indicated by the blue arrows. Obinutuzumab dosing schedule: C1D1, 100 mg; C1D2, 900 mg; C1D8 and 15, 1000 mg; C2-6D1, 1000 mg. C, cycle; D, day; G, GA101/obinutuzumab; PR, partial response; RD1, ramp-up day 1; W, week.
The dose-finding stage explored 2 schedules of drug administration during cycle 1 for TLS risk mitigation: schedule A (venetoclax ramp-up, followed by obinutuzumab) and schedule B (obinutuzumab loading dose over 21 days, followed by venetoclax initiation). Dose escalation occurred according to schedule A in R/R patients before initiating cohorts according to schedule B or any 1L patients. Standard 3 + 3 dose-escalation rules were applied, whereby if no dose-limiting toxicity (DLT) was observed in any 3 patients in a given cohort, the next cohort could begin enrollment without further expansion (supplemental Table 4). Once R/R patients in schedule A had completed the DLT observation window (supplemental Table 5) for cohort 3 (venetoclax 400 mg) or reached the maximum tolerated dose (MTD), whichever occurred first, an internal monitoring committee (IMC) and external scientific overview committee (SOC; composed of CLL experts) were to provide recommendations for the initial cohort dose of venetoclax for schedule B in the R/R population and for schedule A in the 1L population. Subsequent assessment for dose/schedule recommendations were to be provided by the SOC/IMC according to supplemental Figure 2 to determine the venetoclax dose and schedule for safety expansion. Patients previously enrolled into cohorts with lower target doses of venetoclax than the recommended dose for safety expansion were to be allowed to increase the venetoclax dose once the safety-expansion phase of the study started.
Venetoclax-obinutuzumab was administered for 6 cycles, followed by venetoclax monotherapy until disease progression (PD), unacceptable toxicity, or death in R/R patients, or completion of a 1-year fixed treatment duration in 1L patients. In 1L patients, venetoclax could be extended beyond 1 year if there was detectable MRD in the bone marrow (BM) or the patient was not in complete response (CR).
End points
Primary end points were MTD of venetoclax when combined with obinutuzumab, and safety/tolerability (including incidence/type of protocol predefined DLTs, adverse events [AEs] and serious AEs [SAEs], laboratory variables, and vital signs) of the combination in R/R and 1L patients. DLTs (supplemental Table 5) were defined as grade 4 neutropenia, thrombocytopenia, and infusion-related reactions (IRRs); grade 3-4 febrile neutropenia; clinical TLS; or all other nonhematologic grade 3-5 AEs. Efficacy measures (including CR, overall response rate [ORR; CR plus partial response (PR)], duration of response, and PFS) were secondary end points. uMRD rate was an exploratory end point.
Evaluations
Baseline characteristics including cytogenetic aberrations, mutational analysis of immunoglobulin heavy-chain variable region (IGHV) and TP53 genes, serum β-2-microglobulin, and CD38 expression were assessed centrally. Measurable lymph node size assessments (by computed tomography/magnetic resonance imaging) were mandatory before treatment initiation to assess TLS risk (supplemental Table 3) and subsequently to confirm response.
AEs were assessed at every visit and graded according to National Cancer Institute Common Terminology Criteria for Adverse Events v4.0. In patients with cytopenias at baseline, hematologic toxicity was graded according to the National Cancer Institute–Sponsored Working Group (NCI-WG)26 until recovery of peripheral blood (PB) cells following treatment initiation. TLS was classified according to Howard criteria.27 Response assessments were performed by investigators per iwCLL 2008 criteria,25 with BM examination and imaging to confirm response, starting from cycle 2 whenever there was clinical indication of response. PB MRD samples were taken at baseline, cycle 4, any time CR was determined, every 2 to 3 months after the last obinutuzumab dose (1L and R/R), and every 3 months after the last venetoclax dose (1L). BM MRD samples were taken at CR confirmation, and at 3 months after 1 year of treatment. PB and BM MRD analyses were assessed centrally at The Ohio State University using a 5-color flow cytometry assay (uMRD threshold of 1 CLL cell per 104 cells in samples with a minimum of 200 000 leukocytes) following the European Research Initiative on CLL principle.28
Statistical methods
Planned enrollment was ∼90 patients: 3 to 6 for each dose-finding cohort and ≥14 additional patients for each safety-expansion cohort. Safety analyses included all patients receiving ≥1 dose of any study drug. Efficacy analyses included all patients receiving ≥2 cycles of venetoclax-obinutuzumab (as response assessment with BM examination and imaging according to the iwCLL started from end of cycle 2). MRD analyses included all patients who reached the specified landmark time point, plus those discontinuing the study earlier because of AEs, PD, or death (if applicable). At each assessment, the first evaluable PB MRD sample after that time point was used for analysis. Kaplan-Meier methodology was used for time-to-event analyses.29
The first 4 patients with R/R CLL enrolled were subsequently discontinued from the study, shortly after a sponsor-initiated clinical hold in December 2012, due to clinical TLS events in other early-phase venetoclax studies that resulted in extensive changes to all venetoclax CLL protocols, including the present study. Safety data for these patients were reviewed independently and were not included in the final analyses given the different treatment schedule and limited follow-up available. These data are presented in supplemental Table 6.
Results
Patients
Between May 2013 and March 2016, 46 R/R and 32 1L patients were enrolled; 24 R/R and 12 1L patients during dose finding and 22 and 20, respectively, during safety expansion. One patient did not meet the inclusion criteria (corrected QT interval >470 ms) and discontinued prior to receiving any study drugs. Two patients discontinued treatment due to AEs before completing 2 cycles of combination treatment (1 case each of grade 2 lower respiratory infection and grade 3 ulcerative colitis). The R/R safety and efficacy populations therefore comprised 45 and 43 patients, respectively. All 32 1L patients were included in the analyses (supplemental Figure 3). During dose finding, 23 R/R patients and 12 1L patients were safety evaluable. Among them, 22 (16 R/R, 6 1L) received cycle 1 treatment according to schedule A, and 13 (7 R/R, 6 1L) according to schedule B. Data cutoff was 21 May 2018.
Baseline characteristics are reported in Table 1. Fifty-six percent of R/R patients and 71% of 1L patients had creatinine clearance ≥70 mL/min; 56% and 56%, respectively, were ≤65 years old at screening. In the R/R cohort, median number of prior therapies was 2 (range, 1-6); 79% of patients had received fludarabine-based combinations, 21% Bruton tyrosine kinase inhibitors (BTKi’s), and 14% phosphoinositide-3-kinase inhibitors previously. Among patients with samples available for baseline cytogenetic and/or molecular assessment, 77% of R/R and 57% of 1L patients had unmutated IGHV; del(17p) and/or TP53 mutation was present in 55% of R/R and 17% of 1L patients.
Patient demographics and baseline characteristics for the R/R and 1L populations (efficacy population)
| Characteristic | R/R, N = 43 | 1L, N = 32 |
|---|---|---|
| Median age (range), y | 61 (42-80) | 63 (47-73) |
| Male, n (%) | 30 (70) | 20 (63) |
| ECOG PS, n (%) | ||
| 0 | 22 (51) | 16 (50) |
| 1 | 21 (49) | 16 (50) |
| 2 | 0 | 0 |
| Rai stage at screening, n (%) | ||
| 0 | 0 | 0 |
| I/II | 13 (30) | 9 (28) |
| III/IV | 28 (65) | 18 (56) |
| Unknown | 2 (5) | 5 (16) |
| Creatinine clearance, n/N (%) | ||
| <70 mL/min | 19 (44) | 9/31 (29) |
| ≥70 mL/min | 24 (56) | 22/31 (71) |
| Pretreatment TLS risk, n (%)* | ||
| Low | 10 (23) | 2 (6) |
| Medium | 19 (44) | 22 (69) |
| High | 14 (33) | 8 (25) |
| Cytogenetics, n/N (%)† | ||
| del(17p)/TP53 mut‡ | 23/42 (55) | 5/29 (17) |
| del(11q) | 8/42 (19) | 6/29 (21) |
| Trisomy 12 | 2/42 (5) | 6/29 (21) |
| No abnormalities | 3/42 (7) | 1/29 (3) |
| del(13q) | 6/42 (14) | 11/29 (38) |
| TP53 mutation, n/N (%)§ | 18/40 (45) | 5/26 (19) |
| IGHV unmutated, n/N (%) | 26/34 (77) | 16/28 (57) |
| Serum β-2 microglobulin, n (%) | ||
| ≥3.5 mg/mL | 28 (65) | 19 (59) |
| CD38+, n/N (%)|| | 19/33 (58) | 12/25 (48) |
| Median previous therapies, n (range) | 2 (1-6) | NA |
| Prior therapies received, n (%) | ||
| Fludarabine-based treatment | 34 (79) | NA |
| Bendamustine or BR | 12 (28) | NA |
| BTKi | 9 (21) | NA |
| PI3Ki | 6 (14) | NA |
| Characteristic | R/R, N = 43 | 1L, N = 32 |
|---|---|---|
| Median age (range), y | 61 (42-80) | 63 (47-73) |
| Male, n (%) | 30 (70) | 20 (63) |
| ECOG PS, n (%) | ||
| 0 | 22 (51) | 16 (50) |
| 1 | 21 (49) | 16 (50) |
| 2 | 0 | 0 |
| Rai stage at screening, n (%) | ||
| 0 | 0 | 0 |
| I/II | 13 (30) | 9 (28) |
| III/IV | 28 (65) | 18 (56) |
| Unknown | 2 (5) | 5 (16) |
| Creatinine clearance, n/N (%) | ||
| <70 mL/min | 19 (44) | 9/31 (29) |
| ≥70 mL/min | 24 (56) | 22/31 (71) |
| Pretreatment TLS risk, n (%)* | ||
| Low | 10 (23) | 2 (6) |
| Medium | 19 (44) | 22 (69) |
| High | 14 (33) | 8 (25) |
| Cytogenetics, n/N (%)† | ||
| del(17p)/TP53 mut‡ | 23/42 (55) | 5/29 (17) |
| del(11q) | 8/42 (19) | 6/29 (21) |
| Trisomy 12 | 2/42 (5) | 6/29 (21) |
| No abnormalities | 3/42 (7) | 1/29 (3) |
| del(13q) | 6/42 (14) | 11/29 (38) |
| TP53 mutation, n/N (%)§ | 18/40 (45) | 5/26 (19) |
| IGHV unmutated, n/N (%) | 26/34 (77) | 16/28 (57) |
| Serum β-2 microglobulin, n (%) | ||
| ≥3.5 mg/mL | 28 (65) | 19 (59) |
| CD38+, n/N (%)|| | 19/33 (58) | 12/25 (48) |
| Median previous therapies, n (range) | 2 (1-6) | NA |
| Prior therapies received, n (%) | ||
| Fludarabine-based treatment | 34 (79) | NA |
| Bendamustine or BR | 12 (28) | NA |
| BTKi | 9 (21) | NA |
| PI3Ki | 6 (14) | NA |
BTKi, Bruton tyrosine kinase inhibitor; mut, mutated; NA, not applicable; PI3Ki, phosphoinositide 3-kinase inhibitor.
Low risk if largest node <5 cm diameter and absolute lymphocyte count (ALC) <25 × 109/L, medium risk if ALC ≥25 × 109/L or largest nodes ≥5 cm and <10 cm diameter, or high risk if ALC ≥25 × 109/L and largest node ≥5 cm diameter or largest node ≥10 cm diameter.
Fluorescence in situ hybridization (FISH) cutoffs for positivity: del17p >7%; del11q >6%; del13q >5.5%; trisomy 12 >2.5%.
A modified hierarchical model was used to maximize identification of the higher-risk population due to missing samples for cytogenetic assessment. The del(17p)/TP53 mutated subgroup included patients with a 17p deletion by FISH and/or TP53 mutation by next-generation sequencing (NGS).
By NGS. Cutoff for positivity >5%.
Cutoff for positivity >30%.
Treatment exposure
Eighty percent of R/R patients (36 of 45) and all 1L patients (32 of 32) received venetoclax 400 mg per day. Ninety-three percent of R/R patients (42 of 45) and all 1L patients (32 of 32) completed 6 cycles of venetoclax-obinutuzumab. Median venetoclax treatment duration was 789 days (range, 8-1516 days) and 371 days (range, 314-883 days) in the R/R and 1L populations, respectively. Median relative venetoclax dose intensity (supplemental Dose intensity calculation) was 100% (range, 31-100) and 100% (range, 53-100) for R/R and 1L patients, respectively. Twelve 1L patients received venetoclax beyond 1 year (range, 408-883 days).
Safety
During dose finding, R/R patients were enrolled to either the 100-mg (schedule A), 200-mg (schedule A), or 400-mg (schedule A or B) venetoclax dose cohorts. As the 1L cohorts were initiated after the R/R cohort’s safety assessment by the IMC and SOC, all 12 1L patients enrolled during the dose finding were included in the 400-mg (schedule A or B) venetoclax dose cohort. No DLTs (including clinical TLS) were observed with either schedule A or B during dose finding, and the MTD was not reached.
There were no differences in safety, including rate of TLS events between schedules. During dose finding, 4 laboratory grade 3 TLS events were observed in 3 patients (all R/R): 2 events each with schedules A and B. One TLS event with schedule B occurred after obinutuzumab administration but before introduction of venetoclax. The other 3 events (2 with schedule A and 1 with schedule B) occurred during venetoclax ramp-up. Details of TLS events are summarized in supplemental Table 7. After reviewing the safety database of the dose-finding phase and program-wide data, the IMC and SOC recommended schedule B for obinutuzumab debulking followed by venetoclax, and a venetoclax dose of 400 mg for the safety-expansion phase.
All 77 safety-evaluable patients reported ≥1 AE, mostly grade 1-2 (87%). The most frequent AEs (any grade) were infections, diarrhea, IRRs, nausea, and neutropenia (Table 2). Infections were mainly low grade and driven by upper respiratory tract infections and sinusitis (Table 2). Overall, 64% of R/R (29 of 45) and 81% of 1L patients (26 of 32) received treatment according to schedule B and all IRR events were grade 1-2 except for 2 grade 3 events observed in the R/R cohort; none led to obinutuzumab discontinuation.
Treatment-emergent AEs (safety population)
| Patients, n (%) | R/R (N = 45) | 1L (N = 32) | ||
|---|---|---|---|---|
| All grade | Grade 3-4 | All grade | Grade 3-4 | |
| AEs occurring in ≥20% of patients | ||||
| Diarrhea | 31 (69) | 3 (7) | 18 (56) | 1 (3) |
| Infusion-related reaction | 29 (64) | 2 (4) | 22 (69) | 0 |
| Neutropenia | 29 (64) | 26 (58) | 21 (66) | 17 (53) |
| Fatigue | 24 (53) | 1 (2) | 14 (44) | 1 (3) |
| Nausea | 23 (51) | 0 | 22 (69) | 0 |
| Cough | 22 (49) | 0 | 11 (34) | 0 |
| Pyrexia | 20 (44) | 0 | 15 (47) | 1 (3) |
| Anemia | 19 (42) | 2 (4) | 9 (28) | 1 (3) |
| Chills | 16 (36) | 0 | 11 (34) | 0 |
| Thrombocytopenia | 15 (33) | 10 (22) | 14 (44) | 7 (22) |
| Headache | 15 (33) | 0 | 12 (38) | 0 |
| Vomiting | 14 (31) | 1 (2) | 11 (34) | 0 |
| Dyspnea | 12 (27) | 0 | 9 (28) | 0 |
| Arthralgia | 12 (27) | 0 | 3 (9) | 0 |
| Dizziness | 10 (22) | 0 | 6 (19) | 0 |
| Constipation | 9 (20) | 0 | 8 (25) | 0 |
| Hyperphosphatemia | 9 (20) | 1 (2) | 2 (6) | 0 |
| Rash | 8 (18) | 0 | 7 (22) | 0 |
| Abdominal pain | 6 (13) | 0 | 8 (25) | 0 |
| Hypotension | 5 (11) | 0 | 7 (22) | 0 |
| Flushing | 4 (9) | 0 | 10 (31) | 0 |
| Chest discomfort | 4 (9) | 0 | 7 (22) | 0 |
| Dyspepsia | 4 (9) | 0 | 7 (22) | 0 |
| Infection AEs occurring in >5% of patients | ||||
| Infections and infestations (system organ class) | 38 (84) | 13 (29) | 26 (81) | 4 (13) |
| Upper respiratory tract infection | 17 (38) | 1 (2) | 6 (19) | 0 |
| Sinusitis | 12 (27) | 0 | 5 (16) | 0 |
| Pneumonia | 7 (16) | 5 (11) | 1 (3) | 0 |
| Lower respiratory tract infection | 6 (13) | 2 (4) | 2 (6) | 0 |
| Cellulitis | 5 (11) | 4 (9) | 1 (3) | 0 |
| Rhinovirus infection | 5 (11) | 0 | 0 | 0 |
| Urinary tract infection | 4 (9) | 2 (4) | 4 (13) | 0 |
| Influenza | 4 (9) | 1 (2) | 2 (6) | 0 |
| Herpes zoster | 3 (7) | 0 | 1 (3) | 0 |
| Diverticulitis | 2 (4) | 0 | 2 (6) | 1 (3) |
| Bronchitis | 2 (4) | 0 | 3 (9) | 0 |
| Skin infection | 2 (4) | 0 | 2 (6) | 0 |
| Nasopharyngitis | 2 (4) | 0 | 2 (6) | 0 |
| Respiratory syncytial virus infection | 1 (2) | 0 | 2 (6) | 0 |
| Fungal skin infection | 0 | 0 | 2 (6) | 0 |
| Patients, n (%) | R/R (N = 45) | 1L (N = 32) | ||
|---|---|---|---|---|
| All grade | Grade 3-4 | All grade | Grade 3-4 | |
| AEs occurring in ≥20% of patients | ||||
| Diarrhea | 31 (69) | 3 (7) | 18 (56) | 1 (3) |
| Infusion-related reaction | 29 (64) | 2 (4) | 22 (69) | 0 |
| Neutropenia | 29 (64) | 26 (58) | 21 (66) | 17 (53) |
| Fatigue | 24 (53) | 1 (2) | 14 (44) | 1 (3) |
| Nausea | 23 (51) | 0 | 22 (69) | 0 |
| Cough | 22 (49) | 0 | 11 (34) | 0 |
| Pyrexia | 20 (44) | 0 | 15 (47) | 1 (3) |
| Anemia | 19 (42) | 2 (4) | 9 (28) | 1 (3) |
| Chills | 16 (36) | 0 | 11 (34) | 0 |
| Thrombocytopenia | 15 (33) | 10 (22) | 14 (44) | 7 (22) |
| Headache | 15 (33) | 0 | 12 (38) | 0 |
| Vomiting | 14 (31) | 1 (2) | 11 (34) | 0 |
| Dyspnea | 12 (27) | 0 | 9 (28) | 0 |
| Arthralgia | 12 (27) | 0 | 3 (9) | 0 |
| Dizziness | 10 (22) | 0 | 6 (19) | 0 |
| Constipation | 9 (20) | 0 | 8 (25) | 0 |
| Hyperphosphatemia | 9 (20) | 1 (2) | 2 (6) | 0 |
| Rash | 8 (18) | 0 | 7 (22) | 0 |
| Abdominal pain | 6 (13) | 0 | 8 (25) | 0 |
| Hypotension | 5 (11) | 0 | 7 (22) | 0 |
| Flushing | 4 (9) | 0 | 10 (31) | 0 |
| Chest discomfort | 4 (9) | 0 | 7 (22) | 0 |
| Dyspepsia | 4 (9) | 0 | 7 (22) | 0 |
| Infection AEs occurring in >5% of patients | ||||
| Infections and infestations (system organ class) | 38 (84) | 13 (29) | 26 (81) | 4 (13) |
| Upper respiratory tract infection | 17 (38) | 1 (2) | 6 (19) | 0 |
| Sinusitis | 12 (27) | 0 | 5 (16) | 0 |
| Pneumonia | 7 (16) | 5 (11) | 1 (3) | 0 |
| Lower respiratory tract infection | 6 (13) | 2 (4) | 2 (6) | 0 |
| Cellulitis | 5 (11) | 4 (9) | 1 (3) | 0 |
| Rhinovirus infection | 5 (11) | 0 | 0 | 0 |
| Urinary tract infection | 4 (9) | 2 (4) | 4 (13) | 0 |
| Influenza | 4 (9) | 1 (2) | 2 (6) | 0 |
| Herpes zoster | 3 (7) | 0 | 1 (3) | 0 |
| Diverticulitis | 2 (4) | 0 | 2 (6) | 1 (3) |
| Bronchitis | 2 (4) | 0 | 3 (9) | 0 |
| Skin infection | 2 (4) | 0 | 2 (6) | 0 |
| Nasopharyngitis | 2 (4) | 0 | 2 (6) | 0 |
| Respiratory syncytial virus infection | 1 (2) | 0 | 2 (6) | 0 |
| Fungal skin infection | 0 | 0 | 2 (6) | 0 |
Data include all investigator-reported AEs, regardless of relationship to study drug. AEs occurring in ≥20% of patients are listed by Medical Dictionary for Regulatory Activities (MedDRA) preferred term (PT). Infection AEs occurring in >5% patients are listed by MedDRA system organ class and PT.
In the R/R and 1L populations, grade 3-4 AEs were reported in 80% and 78% of patients, respectively, most frequently neutropenia (R/R, 58%; 1L, 53%) (Table 2). Grade 3-4 febrile neutropenia was reported in 16% and 13% of R/R and 1L patients, respectively. The overall rate of grade 3-4 infections was 29% in R/R patients and 13% in 1L patients (Table 2). The most common grade 3-4 infection AEs in R/R patients were pulmonary infection (16%; including preferred terms of pneumonia, lower respiratory tract infection, viral lower respiratory tract infection, and lung infection), cellulitis (9%), and urinary tract infection (4%). Grade 3-4 infections reported in 1L patients included 1 case each of appendicitis, diverticulitis, Enterobacter bacteremia, and viral respiratory tract infection. SAEs were reported in 46% of patients (R/R, 60%; 1L, 34%; supplemental Table 8). During the safety-expansion phase of the study, 1 laboratory TLS event occurred in a 1L patient after obinutuzumab and before initiation of venetoclax (supplemental Table 7). No clinical TLS was reported.
Seventy-six percent of R/R and 66% of 1L patients experienced grade 3-4 AEs during the combination treatment period vs 54% and 34%, respectively, during the venetoclax monotherapy period (supplemental Table 9). Grade 3-4 neutropenia occurred in 48% (R/R, 49%; 1L, 47%) and 24% (R/R, 30%; 1L, 16%) of patients during the combination and monotherapy phases, respectively. In R/R patients, grade 3-4 infections occurred in 18% of patients (8 of 45) during the combination treatment period compared with 21% of patients (9 of 43) during the venetoclax monotherapy period. In the 1L population, 6% of patients (2 of 32) reported grade 3-4 infections in each of the combination and monotherapy periods.
Venetoclax was discontinued due to AEs in 16% of R/R patients (7 of 45) and 3% of 1L patients (1 of 32); most occurred after 1 year of treatment. Obinutuzumab was discontinued due to AEs in 4% of R/R patients (2 of 45) and no 1L patients (Table 3).
Venetoclax and obinutuzumab discontinuations due to AEs
| AEs | Grade | Study day of treatment discontinuation |
|---|---|---|
| AEs leading to venetoclax discontinuation | ||
| R/R cohort | ||
| Diarrhea in context of ulcerative colitis* | 3 | 29 |
| Thrombocytopenia | 2 | 652 |
| Lymphopenia | 3 | |
| Autoimmune hemolytic anemia | 2 | 954 |
| Pneumonia in context of a metastatic squamous cell carcinoma of the lung† | 3 | 619 |
| Fatigue in context of persistent anemia | 2 | 331 |
| Intermittent long-lasting diarrhea | 1 | 575 |
| Esophageal adenocarcinoma | 3 | 722 |
| 1L cohort | ||
| Diarrhea | 3 | 346 |
| AEs leading to obinutuzumab discontinuation | ||
| R/R cohort | ||
| Lower respiratory tract infection | 2 | 29 |
| Ulcerative colitis* | 3 | 43 |
| 1L cohort–no discontinuations due to AEs |
| AEs | Grade | Study day of treatment discontinuation |
|---|---|---|
| AEs leading to venetoclax discontinuation | ||
| R/R cohort | ||
| Diarrhea in context of ulcerative colitis* | 3 | 29 |
| Thrombocytopenia | 2 | 652 |
| Lymphopenia | 3 | |
| Autoimmune hemolytic anemia | 2 | 954 |
| Pneumonia in context of a metastatic squamous cell carcinoma of the lung† | 3 | 619 |
| Fatigue in context of persistent anemia | 2 | 331 |
| Intermittent long-lasting diarrhea | 1 | 575 |
| Esophageal adenocarcinoma | 3 | 722 |
| 1L cohort | ||
| Diarrhea | 3 | 346 |
| AEs leading to obinutuzumab discontinuation | ||
| R/R cohort | ||
| Lower respiratory tract infection | 2 | 29 |
| Ulcerative colitis* | 3 | 43 |
| 1L cohort–no discontinuations due to AEs |
Both AEs occurring in the same patient.
Metastatic squamous cell carcinoma of the lung led to death on day 667 of the study.
Three patients (7%) in the R/R population had a fatal AE (acute respiratory failure in a patient with suspected Richter transformation, pneumonia in the context of metastatic squamous cell carcinoma of the lung, and pneumonia reported ∼3 months following the last venetoclax dose). No deaths were reported in the 1L population.
Efficacy
ORR (best response) was 95% (95% confidence interval [CI], 84-99) and 100% (95% CI, 89-100) in R/R and 1L patients, respectively (Table 4). Thirty-seven percent of R/R patients (95% CI, 23-53) and 78% of 1L patients (95% CI, 60-91) achieved CR/CR with incomplete marrow recovery (CRi) as best response. Responses were similar among patients in the different cytogenetic subgroups (Table 4).
Best response to treatment according to iwCLL 2008 criteria
| Response, n (%) | Entire efficacy population | By cytogenetic abnormalities* | By IGHV status | |||||
|---|---|---|---|---|---|---|---|---|
| del(17p)/TP53 mut | cdel(11q) | Trisomy 12 | None | del(13q) | Mutated | Unmutated | ||
| R/R population, N† | 43 | 16 | 9 | 2 | 4 | 11 | 4 | 26 |
| ORR | 41 (95) | 15 (94) | 8 (89) | 2 (100) | 4 (100) | 11 (100) | 4 (100) | 24 (92) |
| CR/CRi | 16 (37) | 4 (25) | 4 (44) | 2 (100) | 3 (75) | 2 (18) | 4 (100) | 9 (35) |
| PR | 25 (58) | 11 (69) | 4 (44) | 0 | 1 (25) | 9 (82) | 0 | 15 (58) |
| SD | 2 (5) | 1 (6) | 1 (11) | 0 | 0 | 0 | 0 | 2 (8) |
| 1L population, N† | 32 | 5 | 6 | 6 | 1 | 11 | 11 | 16 |
| ORR | 32 (100) | 5 (100) | 6 (100) | 6 (100) | 1 (100) | 11 (100) | 11 (100) | 16 (100) |
| CR/CRi | 25 (78) | 3 (60) | 5 (83) | 5 (83) | 1 (100) | 9 (82) | 10 (91) | 12 (75) |
| PR | 7 (22) | 2 (40) | 1 (17) | 1 (17) | 0 | 2 (18) | 1 (9) | 4 (25) |
| Response, n (%) | Entire efficacy population | By cytogenetic abnormalities* | By IGHV status | |||||
|---|---|---|---|---|---|---|---|---|
| del(17p)/TP53 mut | cdel(11q) | Trisomy 12 | None | del(13q) | Mutated | Unmutated | ||
| R/R population, N† | 43 | 16 | 9 | 2 | 4 | 11 | 4 | 26 |
| ORR | 41 (95) | 15 (94) | 8 (89) | 2 (100) | 4 (100) | 11 (100) | 4 (100) | 24 (92) |
| CR/CRi | 16 (37) | 4 (25) | 4 (44) | 2 (100) | 3 (75) | 2 (18) | 4 (100) | 9 (35) |
| PR | 25 (58) | 11 (69) | 4 (44) | 0 | 1 (25) | 9 (82) | 0 | 15 (58) |
| SD | 2 (5) | 1 (6) | 1 (11) | 0 | 0 | 0 | 0 | 2 (8) |
| 1L population, N† | 32 | 5 | 6 | 6 | 1 | 11 | 11 | 16 |
| ORR | 32 (100) | 5 (100) | 6 (100) | 6 (100) | 1 (100) | 11 (100) | 11 (100) | 16 (100) |
| CR/CRi | 25 (78) | 3 (60) | 5 (83) | 5 (83) | 1 (100) | 9 (82) | 10 (91) | 12 (75) |
| PR | 7 (22) | 2 (40) | 1 (17) | 1 (17) | 0 | 2 (18) | 1 (9) | 4 (25) |
Responses by cytogenetic abnormalities according to hierarchical model.
N for responses according to cytogenetic abnormalities and IGHV status may include only patients with samples available for cytogenetic and IGHV status assessment, respectively.
Rates of PB uMRD were 64% (27 of 42) and 91% (29 of 32) in R/R and 1L patients, respectively, ≥3 months after last obinutuzumab dose. After a median of 12.0 months (range, 11.1-18.4 months) from last obinutuzumab dose, PB uMRD rates were sustained at 63% (25 of 40) in R/R patients (Figure 2A) and 78% (25 of 32) in 1L patients (Figure 2C). For 1L patients, ≥3 months after completion of all treatment (median, 4.4 months [range, 2.8-8.5 months] from last venetoclax dose), the rate of PB uMRD was 72% (23 of 32) (Figure 2D).
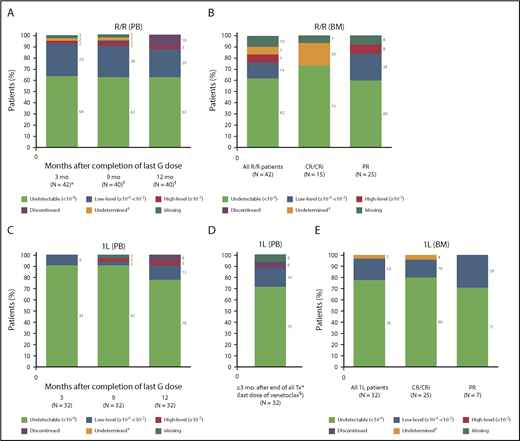
MRD rates in PB and BM. (A) MRD rates in PB according to time after last dose of obinutuzumab in the R/R population. (B) MRD rates in BM by best response achieved in the R/R population. (C) MRD rates in PB according to time after the last dose of obinutuzumab in the 1L population. (D) MRD rates in PB after completion of all treatment (last dose of venetoclax) in the 1L population. (E) MRD rates in BM by best response achieved in the 1L population. “Discontinued” specifies the number of patients who discontinued the study before the landmark time point due to PD, death, or AE (if applicable). “Missing” specifies the number of patients who reached the landmark time point but did not have samples available for MRD assessment. *Of 43 R/R patients included in the efficacy analysis, 1 was excluded from the MRD analysis because of an undetectable MRD result at screening assumed to be due to the use of anti-CD20 <2 months before starting the trial. †Two patients discontinued the study before achieving this time point due to other reasons than PD, death, or AE, and were excluded from the MRD analysis at this landmark time point. ‡Undetermined: <10−4, but <200 000 leukocytes analyzed. §Median 4.4 months (range, 2.8-8.5 months) from last dose of venetoclax. CRi, complete response with incomplete marrow recovery; G, GA101/obinutuzumab; Tx, treatment.
MRD rates in PB and BM. (A) MRD rates in PB according to time after last dose of obinutuzumab in the R/R population. (B) MRD rates in BM by best response achieved in the R/R population. (C) MRD rates in PB according to time after the last dose of obinutuzumab in the 1L population. (D) MRD rates in PB after completion of all treatment (last dose of venetoclax) in the 1L population. (E) MRD rates in BM by best response achieved in the 1L population. “Discontinued” specifies the number of patients who discontinued the study before the landmark time point due to PD, death, or AE (if applicable). “Missing” specifies the number of patients who reached the landmark time point but did not have samples available for MRD assessment. *Of 43 R/R patients included in the efficacy analysis, 1 was excluded from the MRD analysis because of an undetectable MRD result at screening assumed to be due to the use of anti-CD20 <2 months before starting the trial. †Two patients discontinued the study before achieving this time point due to other reasons than PD, death, or AE, and were excluded from the MRD analysis at this landmark time point. ‡Undetermined: <10−4, but <200 000 leukocytes analyzed. §Median 4.4 months (range, 2.8-8.5 months) from last dose of venetoclax. CRi, complete response with incomplete marrow recovery; G, GA101/obinutuzumab; Tx, treatment.
In the R/R and 1L populations, 62% (26 of 42) and 78% (25 of 32) achieved uMRD in BM, respectively (Figure 2B,E). Specifically, 26% (11 of 42) and 63% (20 of 32) of R/R and 1L patients, respectively, were in CR/CRi and had uMRD in BM. Concordance between PB and BM MRD from paired postbaseline samples was high and similar across the R/R (79% [19 of 24]) and 1L (86% [25 of 29]) populations. Therefore, subsequent analysis of MRD kinetics was based on PB MRD.
Patient-level MRD kinetics are shown in Figure 3. Among R/R patients with PB uMRD ≥3 months after the last obinutuzumab dose, 19% (5 of 27) converted to detectable MRD (low level: ≥10−4 to <10−2 or high-level: ≥10−2) in 2 consecutive assessments; 1 had PD at data cutoff. Among 1L patients with PB uMRD after completion of all treatments (median follow-up, 14.4 months [range, 0.5-27.3] post-venetoclax cessation), 34% (10 of 29) converted to positive MRD in 2 consecutive assessments; 5 maintained low-level MRD, 1 became high-level MRD, and 4 became low-level MRD but returned to uMRD at the last MRD assessment, among whom 1 had confirmed PD at data cutoff. No association was observed between cytogenetics and MRD conversion. Median time to first MRD conversion was 196 days (range, 91-554 days) from last venetoclax dose for the 10 1L patients.
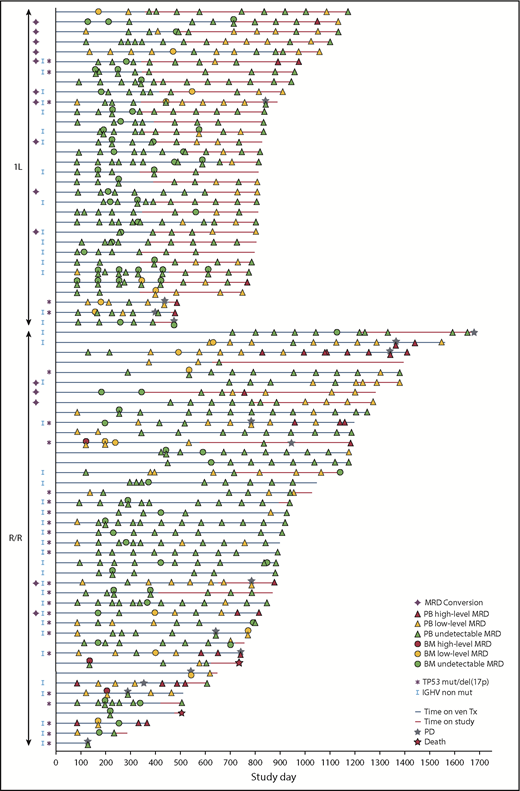
MRD kinetics in individual patients (MRD efficacy-evaluable population). uMRD was defined as <1 CLL cell per 104 mononuclear cells in samples with a minimum of 200 000 leukocytes (<10−4). Low-level MRD was defined as between 1 CLL cell per 104 and 1 cell per 102 mononuclear cells (≥10−4 to <10−2). One patient was excluded from the MRD analysis because of an undetectable MRD result at screening assumed to be due to the use of anti-CD20 <2 months before starting the trial. High-level MRD was defined as ≥1 CLL cell per 102 mononuclear cells (≥10−2). del, deletion; mut, mutated; ven, venetoclax.
MRD kinetics in individual patients (MRD efficacy-evaluable population). uMRD was defined as <1 CLL cell per 104 mononuclear cells in samples with a minimum of 200 000 leukocytes (<10−4). Low-level MRD was defined as between 1 CLL cell per 104 and 1 cell per 102 mononuclear cells (≥10−4 to <10−2). One patient was excluded from the MRD analysis because of an undetectable MRD result at screening assumed to be due to the use of anti-CD20 <2 months before starting the trial. High-level MRD was defined as ≥1 CLL cell per 102 mononuclear cells (≥10−2). del, deletion; mut, mutated; ven, venetoclax.
After a median follow-up of 29.3 months (range, 3-55 months) in R/R patients and 26.7 months (range, 16-39 months) in 1L patients, estimated 24-month PFS was 85.4% (95% CI, 74.5-96.2) and 90.6% (95% CI, 80.5-100), respectively (supplemental Figure 4A-B). Median duration of response was 40.9 months (range, 39.9-51.8 months) and was not reached in R/R and 1L patients, respectively. In total, PD occurred in 16 patients (R/R, n = 12; 1L, n = 4). Of the R/R patients who progressed, 7 of 12 had del(17p)/TP53 mutation at baseline and 7 of 12 were on venetoclax at PD. Three of the 4 1L patients who progressed had del(17p)/TP53 mutation at baseline and 2 were on venetoclax at PD. Richter’s transformation occurred in 1 R/R patient (diffuse large B-cell lymphoma [DLBCL]) and 2 1L patients (DLBCL and Hodgkin lymphoma; the DLBCL case was also MRD+ for CLL in PB and BM; the Hodgkin lymphoma case was uMRD for CLL in both PB and BM; both events were diagnosed after 1 year on study).
Discussion
The current study established venetoclax 400 mg in combination with obinutuzumab as the dose for the safety-expansion cohorts in the R/R and 1L CLL populations. In both populations, the median age (R/R, 61 years; 1L, 63 years) was lower than that usually associated with the initial diagnosis of CLL (range, 65-70 years).30-32 Dosing schedules assessed were safe and acceptable, with schedule B recommended for the safety expansion. The safety profile of venetoclax-rituximab has recently been established for rituximab initiation after venetoclax ramp-up, using a dosing schedule akin to schedule A.16 Hence, both schedules appear feasible for venetoclax administration with an anti-CD20 antibody.
Venetoclax-obinutuzumab had an acceptable safety profile, with expected and manageable toxicities. Most patients completed the planned treatment regimen. The absence of clinical TLS and low incidence of laboratory TLS with venetoclax supported the effectiveness of the ramp-up and prophylactic management strategy.
In the R/R and 1L populations, most AEs were low grade and, as expected based on the known modes of action and established tolerability profiles of both study drugs,19,21,33 neutropenia was the most common grade 3-4 AE. Neutropenia was manageable with standard-of-care measures, and did not result in complications. Grade 3-4 neutropenia was more frequent during combination therapy than monotherapy, confirming tolerability of single-agent venetoclax. Rates of grade 3-4 infections were not especially high and were as expected for this combination and patient population, with no increased mortality due to infection. Although the emergence of hematologic toxicities with this regimen was higher than observed with BTKi’s such as ibrutinib,34 the incidence of grade 3-4 neutropenia (R/R, 58%; 1L, 53%) was lower than seen with fludarabine-cyclophosphamide plus rituximab (FCR) in 1L patients in the CLL10 trial35 and similar to that observed with venetoclax-rituximab in the MURANO study (58% in R/R patients).16 Furthermore, in 1L patients, the incidence of grade 3-4 infections (13%) was lower than seen with chemoimmunotherapy in CLL10 (FCR, 39%; BR, 25%)35 and similar to that seen with ibrutinib (13%).36 In R/R patients, the grade 3-4 infection rate (29%) was higher than seen with venetoclax-rituximab in MURANO (18%)16 and lower than seen with ibrutinib (51%).36 However, direct comparisons are difficult given differences in sample sizes, baseline characteristics, treatment duration, and follow-up between studies.
Despite the study population including a high proportion of patients with poor cytogenetics, all patients except 2 with R/R CLL (both with bulky disease and del[17p] or del[11q]) responded to treatment, demonstrating venetoclax-obinutuzumab as an efficacious regimen in R/R and 1L CLL. Importantly, high response rates and deep remissions were observed in most patient subgroups regardless of cytogenetics and/or physical fitness. PFS in both populations was promising but must be viewed in the context of a small phase 1 study. We did not observe any association between cytogenetics and MRD conversion.
uMRD was sustained at 63% ≥1 year from last obinutuzumab dose in R/R patients and at 72% after completion of all treatment in 1L patients. High uMRD rates ≥1 year after cessation of obinutuzumab allays concerns regarding MRD− status during obinutuzumab treatment. Furthermore, the high rate of uMRD in BM, and the concordance between BM and PB MRD data, suggest that PB MRD could predict BM MRD status in patients treated with venetoclax-obinutuzumab.
MRD status is a known predictor of PFS with chemoimmunotherapy.20,23,35,36 The deep remission rates we observed with venetoclax-obinutuzumab have not been reported with previously available CLL treatments, including FCR, which is currently considered the most efficacious regimen with limited-duration therapy.35,37 MURANO also demonstrated high uMRD rates and the value of uMRD in predicting improved outcome for a fixed-duration, chemotherapy-free regimen with venetoclax-rituximab.17 Therefore, it is expected the high, sustained rate of uMRD seen with venetoclax-obinutuzumab would lead to improved outcomes.
Reemergence of MRD positivity, mainly low-level, was observed in 5 R/R and 10 1L patients; of the 1L patients, only 1 had PD, thereby indicating the potential feasibility of time-limited therapy. Longer follow-up and larger trials are needed to explore the predictive value of deep remissions (CR with uMRD) and the impact of MRD conversion on the appearance of clinical progression using time-limited therapy.
Ongoing studies are investigating venetoclax-obinutuzumab and other venetoclax combinations as doublets or triplets in CLL, including a phase 1b/2 study of venetoclax-obinutuzumab-ibrutinib.38 Preliminary results for 12 patients with R/R CLL showed a high ORR (92%) and deep remissions (all 12 patients became uMRD in PB or BM), consistent with our findings for venetoclax-obinutuzumab; however, longer follow-up is needed to determine longer-term outcomes.38 Ibrutinib-venetoclax has also shown promising clinical activity in the frontline treatment of CLL in early data from the phase 2 CAPTIVATE study.39 In the first 30 patients, uMRD in PB was reached by 77%. These results are consistent with those achieved in our study, which showed similar rates with 1-year limited-duration treatment in 1L patients. Furthermore, although longer follow-up is needed, and with the caveat of the small population size, the CR and uMRD rates observed in our study compare favorably with the results of a phase 3 trial (iLLUMINATE) in the frontline CLL setting with ibrutinib-obinutuzumab,40 in which CR and uMRD (PB or BM) were achieved by 41% and 35% of patients, respectively.
Different approaches to deliver efficacious venetoclax-obinutuzumab are under investigation. Recently, preliminary results of a phase 2 study with bendamustine debulking followed by venetoclax-obinutuzumab showed high ORRs and deep remissions in all subgroups of CLL patients, regardless of whether patients completed the planned 2 cycles of bendamustine debulking.41 Bendamustine debulking contributed to normalization of the lymphocyte count so that the risk category for development of TLS could be downgraded before initiation of venetoclax-obinutuzumab. No incidences of clinical TLS were reported. Additionally, preliminary results from the first 30 patients enrolled in the HOVON 139/GIVE trial, in which 2 cycles of obinutuzumab were given for debulking before venetoclax-obinutuzumab, reported no incidences of clinical TLS, and only 4 patients had laboratory TLS.42 This study also demonstrated early signs of efficacy with venetoclax-obinutuzumab (including high rates of uMRD) in 1L unfit patients.
Our results confirm favorable benefit-risk for R/R and 1L CLL patients at the established dose of 400 mg of venetoclax together with the standard dose of obinutuzumab. For 1L-fit patients, a phase 3 study of venetoclax-obinutuzumab vs chemoimmunotherapy (FCR or BR) vs triplet therapy with ibrutinib is ongoing (CLL13; NCT02950051). In 1L unfit CLL, results from the safety run-in of the phase 3 CLL14 study, using venetoclax-obinutuzumab in patients with 1L CLL and coexisting medical conditions, similarly showed an acceptable safety profile, high uMRD rates, and promising PFS.43 If the primary end points of these large-scale trials are met, venetoclax-obinutuzumab may become a new standard treatment option in 1L CLL irrespective of clinical fitness.
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (www.clinicalstudydatarequest.com). Further details on Roche’s criteria for eligible studies are available here: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here: https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
Previous interim analyses of this study were presented orally at the 57th and 59th annual meetings of the American Society of Hematology, Orlando, FL, 5-8 December 2015, and Atlanta, GA, 9-12 December 2017, respectively.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors give special thanks to the patients and their families, investigators, study coordinators, and support staff, and all Genentech GP28331 study team members. The authors acknowledge Elizabeth A. Punnoose for technical evaluation, and Richa Rajwanshi for safety data interpretation. Venetoclax is being developed in collaboration between Genentech and AbbVie. Tara Miller of Envision Pharma Group and Susan Hasmall and Kate Rijnen of Gardiner-Caldwell Communications provided writing and editorial assistance based on specific direction from the authors; this service was funded by F. Hoffmann-La Roche Ltd.
This work was supported by Genentech Inc and AbbVie. Genentech and AbbVie participated in the design, study conduct, and data analysis and interpretation.
Authorship
Contribution: I.W.F. designed the study, collected, assembled, analyzed, and interpreted data, participated in the writing process and manuscript development, and gave final approval; J.G.G. designed the study, analyzed and interpreted data, participated in the writing process and manuscript development, gave final approval, and was a member of the Scientific Overview Committee; M.J.S.D., W.W., A.Q.-M., L.Y., H.S.W., and Y.J. collected, assembled, analyzed, and interpreted data, participated in the writing process and manuscript development, and gave final approval; M.B.M., R.R.F., P.H., K.A.R., S.P.I., M.V., H.H., and K.H. analyzed and interpreted data, participated in the writing process and manuscript development, and gave final approval; C.K. and T.J.K. designed the study, analyzed and interpreted data, participated in the writing process and manuscript development, and gave final approval; G.L. generated, collected, interpreted, and assembled CLL MRD data, participated in the writing process and manuscript development, and gave final approval; D.S.P. analyzed and interpreted data and participated in the writing process; and M.M. generated, collected, interpreted, assembled, analyzed, and interpreted data, participated in the writing process and manuscript development, and gave final approval.
Conflict-of-interest disclosure: I.W.F. has received research funding for his institution from AbbVie, Acerta, Agios, ArQule, Beigene, Calithera, Celgene, Constellation, Curis, Forma, Forty Seven, Genentech, Gilead, Incyte, Infinity, Janssen, KITE, Merck, Novartis, Pfizer, Pharmacyclics, Portola, Seattle Genetics, Takeda, TG Therapeutics, Trillium, and Verastem. J.G.G. has received honoraria from Roche, AbbVie, Pharmacyclics, Celgene, and Janssen; has held a consulting or advisory role for Roche, Acerta, AbbVie, Pharmacyclics, Celgene, and Janssen; and has received research funding from Celgene, Acerta, and Janssen. M.J.S.D. has received honoraria from, and has held a consulting or advisory role for, AbbVie and Roche, and has received travel, accommodation, or expenses from AbbVie; his institution has received research funding from Roche. W.W. has received travel, accommodation, or expenses from AbbVie, Genentech/Roche, Janssen, Pharmacyclics, Gilead, and Pfizer; his institution has received research funding from GlaxoSmithKline/Novartis, AbbVie, Genentech, Karyopharm, Pharmacyclics LLC, Acerta, Gilead Sciences, Juno Therapeutics, KITE Pharma, Sunesis, Miragen, Oncternal Therapeutics, Inc, Cyclacel, Loxo Oncology, Janssen, and Xencor. R.R.F. has received honoraria from Janssen and Genentech; has held a consulting or advisory role for AbbVie, Genentech, Gilead, Janssen, Acerta/AstraZeneca, Pharmacyclics, Sunesis, Loxo Oncology, TG Therapeutics, and Verastem; has received research funding from Acerta and TG Therapeutics; and has served on a data safety monitoring board for Incyte. P.H. has received honoraria from, and participated in speakers’ bureaus for, Janssen, AbbVie, and Gilead, and has received travel, accommodation, or expenses from Janssen and AbbVie; his institution has received research funding from Pharmacyclics, Janssen, AbbVie, Gilead, and Roche. K.A.R. has held a consulting or advisory role for Acerta and has received research funding from Genentech. S.P.I. has held a consulting or advisory role for Genentech, Takeda, and Bristol-Myers Squibb, and has received research funding from Genentech, Takeda, Spectrum Pharmaceuticals, Seattle Genetics, Bristol-Myers Squibb, Amgen, and Rhizem. A.Q.-M. has received research funding from Roche and Glycart. L.Y. has held a consulting or advisory role for Janssen, Roche, Gilead, and AbbVie, and has received research funding from Roche and Janssen. H.S.W. has received honoraria from Gilead and AbbVie; has held a consulting or advisory role for AbbVie; and has received travel, accommodation, or expenses from AbbVie and Gilead. M.V. is an employee of AbbVie. C.K. and an immediate family member of C.K. are employees of Roche and own Roche stock; C.K. also has a patent or intellectual property interest to declare from Roche. H.H. is an employee of Roche. Y.J. is an employee of Genentech and owns Roche stock. G.L. has research funding to disclose from Genentech, Boehringer Ingelheim, Stemline, Celgene, and BC Pharma. D.S.P. and K.H. are employees of Roche and own Roche stock. M.M. is an employee of Genentech and holds Roche stock. T.J.K. is an employee of UC San Diego Health and Moores Cancer Center; has held a consulting or advisory role for Gilead, Celgene, Roche/Genentech, AbbVie, and Pharmacyclics; has received honoraria from Gilead, AbbVie, Pharmacyclics, Janssen, and Verastem; and has received research funding from Roche/Genentech, AbbVie, Pharmacyclics, and Oncternal. M.B.M. declares no competing financial interests.
Correspondence: Ian W. Flinn, Sarah Cannon Research Institute/Tennessee Oncology, 250 25th Ave North, Suite 412, Nashville, TN 37203; e-mail: iflinn@tnonc.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal