

TO THE EDITOR:
Most patients with chronic lymphocytic leukemia (CLL) are elderly (70% are older than age 65 years1 ) and unfit, often with 1 or more comorbidities, thus making them ineligible for intensive chemoimmunotherapy regimens such as rituximab plus fludarabine and cyclophosphamide.2 The pivotal CLL11 trial (NCT01010061) established obinutuzumab (GA101 [G]; a type II, glycoengineered, anti-CD20 monoclonal antibody with enhanced antibody-dependent cellular cytotoxicity and improved direct B-cell killing3-5 ) plus chlorambucil (Clb) as a new standard of care for previously untreated CLL patients with comorbidities.6,7 In CLL11, G-Clb demonstrated a greater effect on progression-free survival (PFS) and overall survival (OS) vs rituximab plus chlorambucil (R-Clb) and vs Clb alone.8-10
PFS is a standard primary end point in phase 3 CLL trials; however, as advances in treatment options continue to evolve, longer follow-up periods (up to 5 years) are being required to reach this end point, and shorter-term end points are consequently being sought. Measurement of minimal residual disease (MRD) has been identified as a potential surrogate marker for PFS (and possibly OS),11,12 and may provide earlier information on the effectiveness of new treatment strategies in CLL trials. Previous studies have shown that post-induction MRD levels can independently predict PFS13-16 ; however, most data have been generated in young, physically fit patients.
The objectives of the current analysis were to prospectively investigate the relative effect of treatment with G-Clb vs R-Clb on MRD levels and to explore the prognostic value of MRD assessment in patients with previously untreated CLL and comorbidities enrolled in the CLL11 study. CLL11 was an open-label, randomized, 3-arm, phase 3 study that evaluated the efficacy and safety of G-Clb and R-Clb vs Clb alone (stage 1) and G-Clb vs R-Clb (stage 2), in patients with previously untreated CLL and comorbidities (supplemental Figure 1, available on the Blood Web site).8,9 Only patients from stage 2 (data cutoff, October 2017) are considered here. CLL11 was conducted in accordance with the Declaration of Helsinki and was approved by the institutional review board or independent ethics committee of each individual institution.
Eligible patients had previously untreated CD20+ CLL (diagnosed according to International Workshop on CLL criteria),17 a Cumulative Illness Rating Scale (CIRS) score of >6 indicating a burden of comorbidities, and/or reduced renal function (creatinine clearance of 30-69 mL/min). Patients were randomly assigned 1:2:2 to receive six 28-day cycles of Clb alone, G-Clb, or R-Clb (see supplemental Data for dosing regimens). Further details on the study design and eligibility criteria have been published elsewhere.8,9
MRD was analyzed prospectively in peripheral blood (PB) and bone marrow at 2 central laboratories in Kiel, Germany, and Rotterdam, the Netherlands (stage 2 analysis). PB samples were taken at repeated time points before, during, and up to 12 months after treatment. MRD values were obtained by polymerase chain reaction (see supplemental Data for full details). Only PB samples taken at the end of treatment (EOT) are considered in this report (additional analyses are described in the supplemental Data).
Patients were classified into 1 of 3 MRD categories: MRD positive (≥1% or ≥10−2 [≥100 CLL cells per 10 000 leukocytes]); MRD intermediate (<1% and ≥0.01% or <10−2 and ≥10−4 [1-99 CLL cells per 10 000 leukocytes]); or MRD undetectable (<0.01% or <10−4 [<1 CLL cell per 10 000 leukocytes]).14 Patients were included in the population that was evaluable for MRD if they had an MRD sample measurable in PB and/or bone marrow at EOT (within 56 to 190 days of the last day of treatment). Patients with no available MRD sample at EOT but with progressive disease or death within this time frame were considered MRD positive at EOT. Statistical analyses are presented in the supplemental Data.
In total, 781 patients were enrolled; 663 (G-Clb, n = 333; R-Clb, n = 330) completed stage 2. Of these patients, 474 (71.4%) had evaluable PB samples at EOT. Median follow-up was 65.6 months (range, 4.6-85.1 months). Median age was 73 years (range, 39-90 years), with 61.5% of patients (n = 297) age >70 years (supplemental Table 1).
In PB at EOT, 90 patients (19.0%) were categorized as MRD undetectable, 132 (27.8%) as MRD intermediate, and 252 (53.2%) as MRD positive (including 15 patients [3.2%] with progressive disease; 12 patients [2.5%] died). MRD response was significantly associated with PFS (Figure 1A); patients with undetectable MRD had a median PFS of 56.4 months compared with 23.9 months for patients categorized as MRD intermediate (MRD intermediate vs MRD undetectable: hazard ratio [HR], 2.65; 95% confidence interval [CI], 1.91-3.69; P < .001) and 13.9 months for MRD-positive patients (MRD positive vs MRD undetectable: HR, 6.53; 95% CI, 4.78-8.92; P < .001; supplemental Table 2). MRD response was also significantly associated with OS. Median OS was not reached in the undetectable and intermediate MRD categories and was 60.0 months for MRD-positive patients (MRD intermediate vs MRD undetectable: HR, 1.43; 95% CI, 0.91-2.26; P = .125; MRD positive vs MRD undetectable: HR, 2.24; 95% CI, 1.49-3.37; P < .001; supplemental Figure 2).
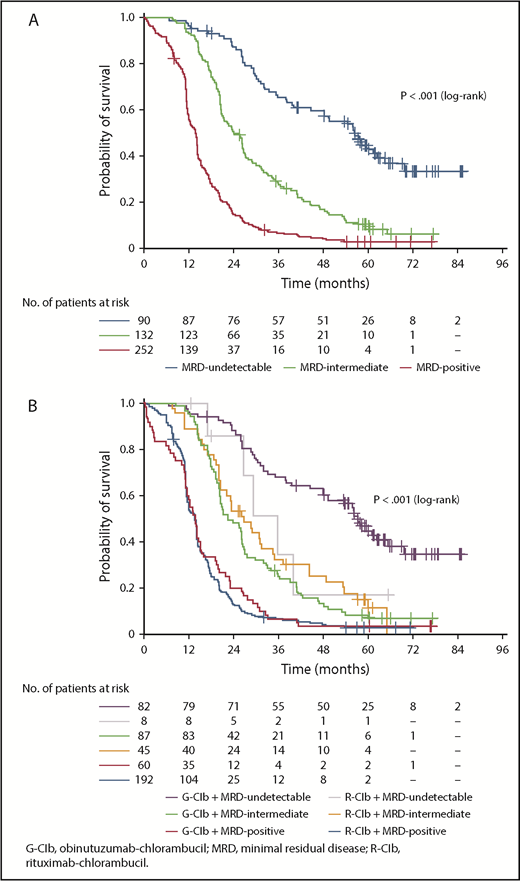
PFS according to MRD category and treatment arm. (A) PFS according to MRD category at EOT in PB; (B) PFS according to MRD category at EOT in PB plus treatment arm.
PFS according to MRD category and treatment arm. (A) PFS according to MRD category at EOT in PB; (B) PFS according to MRD category at EOT in PB plus treatment arm.
Undetectable MRD in PB at EOT was significantly more common in patients receiving G-Clb vs those receiving R-Clb (35.8% vs 3.3%, respectively; P < .001; supplemental Table 3). For patients categorized as MRD undetectable, PFS was numerically longer in those treated with G-Clb (median, 57.3 months; n = 82) than in those treated with R-Clb (median, 35.6 months; n = 8), although this difference was not statistically significant (P = .120 [log-rank test]; Figure 1B). This observed difference may suggest preferential clearing of PB in the G-Clb arm; however, the numbers of patients in the R-Clb arm are too low to allow any firm conclusions to be drawn. Median PFS was comparable for MRD-intermediate patients across arms, and no between-treatment difference was observed for MRD-positive patients (Figure 1B).
The following MRD response categories were used as variables in the multivariable analysis: MRD positive, which included all patients with detectable levels of MRD (ie, all those previously categorized as MRD positive or MRD intermediate), and MRD undetectable. After multivariable analysis, MRD positivity was identified as an independent prognostic factor for PFS in PB at EOT (HR, 3.94; 95% CI, 2.75-5.64; P < .001), as was treatment with G-Clb (HR, 0.58; 95% CI, 0.46-0.73; P < .001). Other independent prognostic factors for PFS were serum thymidine kinase >10 U/L and the presence of genetic risk factors (Table 1). MRD positivity in PB at EOT was also identified as an independent prognostic factor for OS (HR, 1.98; 95% CI, 1.26-3.12; P < .003; supplemental Table 4).
Multivariable analysis of the effects of prognostic factors on PFS in combination with MRD categories in PB at EOT
| Cox regression | Univariable comparison | HR | 95% CI | P |
|---|---|---|---|---|
| Treatment arm | ||||
| G-Clb | vs R-Clb | 0.58 | 0.46-0.73 | <.001 |
| MRD in PB | ||||
| Positive/progressive disease/death | vs undetectable | 3.94 | 2.75-5.64 | <.001 |
| White blood cell count | ||||
| ≥50 | vs <50 | 1.33 | 1.05-1.69 | .020 |
| Serum thymidine kinase (U/L) | ||||
| >10 | vs ≤10 | 1.35 | 1.09-1.68 | .007 |
| Deletion in 17p and/or TP53 mutation | ||||
| Yes | vs no | 2.70 | 1.91-3.82 | <.001 |
| Deletion in 11q | ||||
| Yes | vs no | 1.87 | 1.43-2.44 | <.001 |
| IGHV mutational status | ||||
| Unmutated | vs mutated | 1.97 | 1.52-2.55 | <.001 |
| Cox regression | Univariable comparison | HR | 95% CI | P |
|---|---|---|---|---|
| Treatment arm | ||||
| G-Clb | vs R-Clb | 0.58 | 0.46-0.73 | <.001 |
| MRD in PB | ||||
| Positive/progressive disease/death | vs undetectable | 3.94 | 2.75-5.64 | <.001 |
| White blood cell count | ||||
| ≥50 | vs <50 | 1.33 | 1.05-1.69 | .020 |
| Serum thymidine kinase (U/L) | ||||
| >10 | vs ≤10 | 1.35 | 1.09-1.68 | .007 |
| Deletion in 17p and/or TP53 mutation | ||||
| Yes | vs no | 2.70 | 1.91-3.82 | <.001 |
| Deletion in 11q | ||||
| Yes | vs no | 1.87 | 1.43-2.44 | <.001 |
| IGHV mutational status | ||||
| Unmutated | vs mutated | 1.97 | 1.52-2.55 | <.001 |
Other variables significantly associated with PFS in univariable analyses (significance level set at 5%) and considered as candidates for the multivariable modeling but not included in the final model are Binet stage and serum β2-microglobulin (mg/L).
IGHV, immunoglobulin heavy chain variable.
There is now increasing evidence to support the use of MRD as a surrogate end point for long-term outcome in clinical trials.18,19 Although most of this evidence comes from studies carried out in young, fit patients,14,20 our data show that MRD retains its prognostic significance in a patient population that is generally older, less fit, and treated with less intense treatment regimens. In addition to MRD and treatment arm, several other risk factors were prognostic for either PFS and/or OS, including Binet stage C, CIRS score, serum thymidine kinase, and genetic risk factors, as also seen previously.21-23
In conclusion, this analysis has shown that MRD status is independently associated with PFS and OS in CLL patients with comorbidities and has the potential to act as a surrogate marker for outcome in clinical trials. In addition, it has confirmed the superiority of G-Clb over R-Clb; G-Clb enables more patients to achieve undetectable levels of MRD.
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (www.clinicalstudydatarequest.com). Further details on Roche’s criteria for eligible studies are available at https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and their families and acknowledge the scientists and technicians of the German CLL Study Group laboratories for the MRD sample assessments and statistical analyses and the Roche CLL11 study team.
This study was supported by F. Hoffmann-La Roche Ltd. Helen Cathro of Gardiner-Caldwell Communications (Macclesfield, United Kingdom) provided medical writing support (under the direction of A.W.L.) and was funded by F. Hoffmann-La Roche Ltd.
Authorship
Contribution: K.H., M.K., M.R., and S.S. contributed to the study design and data acquisition, analysis, and interpretation; J.B., G.F.-R., M.T., and S.R. contributed to the data analysis and interpretation; M.S. and S.P.M. contributed to data acquisition and interpretation; V.G., K.F., and K.T. contributed to the study design and data interpretation; A.W.L., S.B., and U.J.M.M. contributed to data acquisition, analysis, and interpretation; M.B. contributed to data analysis; M.H. and J.J.M.v.D. contributed to data interpretation; and all authors helped write the manuscript and approved the final version for publication.
Conflict-of-interest disclosure: A.W.L. received research funding from Roche and was an advisory board member for AbbVie. V.G. received consulting fees from Roche; was an advisory board member for Roche, Janssen, Gilead Sciences, and AbbVie; and received honoraria from Roche, Janssen, and Gilead Sciences. K.F. received travel grants from Roche. M.S. received consulting fees, research funding, and honoraria from and was an advisory board member for Roche, Janssen, Gilead Sciences, and AbbVie. M.T. received consulting fees and honoraria from Roche, Celgene, Janssen, AbbVie, Bristol-Myers Squibb, Takeda, and Gilead Sciences and research funding from Roche and Celgene. U.J.M.M. received honoraria from and was an advisory board member for Roche. S.S. received consulting fees, speaker honoraria, research funding, and travel support from and was an advisory board member for AbbVie, Celgene, Roche/Genentech, Gilead Sciences, GlaxoSmithKline, Janssen, and Novartis. S.B. received research funding from Roche, Celgene, Janssen, and AbbVie; was an advisory board member for Roche; and received honoraria from Roche, AbbVie, Novartis, and Janssen. M.B. received consulting fees from PRMA, research funding from Amgen, and honoraria from Pfizer and Amgen and was an advisory board member for Incyte and Amgen. M.K. received research funding from Roche. J.J.M.v.D. received research funding from Roche, Amgen, and BD Biosciences. M.H. received honoraria and research funding from and was an advisory board member for AbbVie, Amgen, Celgene, Roche, Gilead Sciences, Janssen, and Mundipharma. M.R. received research funding from and was an advisory board member for Roche. K.T., G.F.-R., and K.H. are employed by F. Hoffmann-La Roche Ltd. The remaining authors declare no competing financial interests.
Correspondence: Anton W. Langerak, Department of Immunology, Laboratory for Medical Immunology, Erasmus University Medical Center, Wytemaweg 80, Rotterdam 3015 CN, The Netherlands; e-mail: a.langerak@erasmusmc.nl.
