

Key Points
Decreased numbers and/or impaired function of natural Treg cells participates in the pathogenesis of ITP.
Low-dose chidamide restored immune tolerance in ITP via modulation of natural Treg cells and CTLA4 gene expression.

Abstract
Increased macrophage phagocytosis of antibody-coated platelets, as well as decreased numbers and/or impaired function of CD4+CD25+Foxp3+ regulatory T (Treg) cells, has been shown to participate in the pathogenesis of immune thrombocytopenia (ITP). Low-dose histone deacetylase inhibitors (HDACi’s) are anti-inflammatory and immunomodulatory agents that can enhance immunosuppression in graft-versus-host disease by increasing the number and function of Foxp3+ Treg cells, but it is unclear whether they have the potential to promote immune tolerance and platelet release in ITP. In this study, we performed in vitro and in vivo experiments and found that a low-dose HDACi (chidamide) alleviated thrombocytopenia in passive and active murine models of ITP. Further, low-dose HDACi’s attenuated macrophage phagocytosis of antibody-coated platelets, stimulated the production of natural Foxp3+ Treg cells, promoted the peripheral conversion of T cells into Treg cells, and restored Treg cell suppression in vivo and in vitro. Finally, we confirmed that low-dose HDACi’s could regulate CTLA4 expression in peripheral blood mononuclear cells through modulation of histone H3K27 acetylation. Low-dose HDACi treatment in ITP could be offset by blocking the effect of CTLA4. Therefore, we propose that low-dose chidamide administration has potential as a novel treatment for ITP in the clinic.
Introduction
Immune thrombocytopenia (ITP) is a common autoimmune bleeding disorder that is characterized by persistent thrombocytopenia. Natural CD4+CD25+Foxp3+ regulatory T (Treg) cells contribute to the maintenance of peripheral immune tolerance, and their defects are thought to play a role in the pathogenesis of ITP.1
Recently, histone deacetylase inhibitors (HDACi’s) have been used for their anti-inflammatory and immunomodulatory activities.2 Low concentrations of HDACi’s reduce inflammation, autoimmune responses, and cytokine production in humans with juvenile idiopathic arthritis,3 as well as in animal models of inflammatory bowel disease and arthritis.4 HDACi’s can also alleviate graft-versus-host disease by enhancing the number and function of Foxp3+ Treg cells.5,6
In this study, low-dose HDACi’s alleviated thrombocytopenia in passive and active murine models of ITP. Furthermore, they attenuated macrophage phagocytosis of antibody-coated platelets, stimulated the production of natural Foxp3+ Treg cells, promoted the peripheral conversion of T cells into Treg cells, and restored Treg cell suppressive function in vivo and in vitro. Finally, low-dose HDACi’s regulated CTLA4 expression in peripheral blood mononuclear cells (PBMCs). Low-dose HDACi treatment in ITP could be offset by blocking the effect of CTLA4. We then performed a chromatin immunoprecipitation sequencing (ChIP-Seq) test and found reduced histone H3K27 acetylation in ITP patients compared with controls. We confirmed that low-dose HDACi’s regulated CTLA4 expression through modulation of histone H3K27 acetylation and had the potential to change the prognosis of ITP patients.
Methods
Patients and controls
Thirty-two patients (17 females and 15 males; age range, 22-72 years; median age, 42 years; platelet count range, 3-36 × 109/L; median platelet count, 17 × 109/L) were enrolled in this study, all of whom were free from any ITP-specific therapy for ≥1 month (Table 1). All patients were diagnosed based on previously published criteria for ITP.7 The healthy control group included 20 healthy adult volunteers (11 females and 9 males; age range, 21-60 years; median age, 35 years; platelet count range, 129-284 × 109/L; median platelet count, 178 × 109/L). Enrollment took place between July of 2014 and July of 2018 at the Department of Hematology, Qilu Hospital, Shandong University and at the Department of Hematology, Jinan Central Hospital affiliated to Shandong University. This study was approved by the Medical Ethical Committees of Qilu Hospital, Shandong University and Jinan Central Hospital affiliated to Shandong University. The study was conducted in accordance with the Declaration of Helsinki.
ITP patient characteristics
| Patient no. | Age, y | Sex | Platelet count, ×109/L | Antiplatelet antibody target | Major previous drugs | Bleeding symptoms | |
|---|---|---|---|---|---|---|---|
| GPIIb/IIIa | GPIb/IX | ||||||
| 1 | 41 | F | 12 | + | − | Steroid, VCR | EC, EP |
| 2 | 46 | M | 18 | + | − | Steroid, IVIg, eltrombopag | EC |
| 3 | 22 | F | 6 | − | + | Steroid, RTX | EC, GH |
| 4 | 37 | F | 25 | + | − | Steroid, rhTPO | EC, PT |
| 5 | 28 | M | 22 | + | − | Steroid, rhTPO, IVIg | EC, PT |
| 6 | 33 | M | 5 | + | + | Steroid, rhTPO, RTX | EC, GH |
| 7 | 45 | F | 32 | − | − | Steroid, IVIg, rhTPO, RTX | PT |
| 8 | 52 | F | 14 | − | + | Steroid, VCR | PT, EC |
| 9 | 35 | F | 20 | + | − | Steroid, RTX, eltrombopag | EC |
| 10 | 60 | M | 31 | + | − | Steroid, rhTPO | GH |
| 11 | 40 | F | 19 | − | − | IVIg, steroid | EC |
| 12 | 23 | M | 17 | + | − | Steroid, RTX | EC |
| 13 | 65 | F | 21 | − | + | IVIg, steroid, rhTPO | EP |
| 14 | 47 | M | 16 | + | − | Steroid, RTX, danazol | GH, EP |
| 15 | 29 | F | 8 | + | + | Steroid, RTX | GUH, EP |
| 16 | 25 | M | 23 | − | − | Steroid, IVIg, rhTPO, RTX | PT |
| 17 | 32 | M | 3 | + | + | Steroid, IVIg, rhTPO, RTX | EC, PT, GH |
| 18 | 42 | F | 15 | − | + | Steroid, danazol, RTX | PT |
| 19 | 38 | F | 8 | + | − | Steroid, splenectomy, RTX | EP |
| 20 | 50 | M | 10 | + | − | IVIg, rhTPO, steroid | PT |
| 21 | 57 | M | 7 | + | − | Steroid, thalidomide, CsA | EC, PT |
| 22 | 40 | F | 11 | − | + | Steroid, VCR | EC, GH |
| 23 | 72 | F | 23 | + | − | Steroid | GH |
| 24 | 45 | M | 36 | − | − | Steroid, RTX | EC |
| 25 | 59 | F | 12 | − | + | None | EC, PT |
| 26 | 48 | F | 19 | + | − | Steroid, RTX, rhTPO | EP |
| 27 | 55 | M | 22 | − | − | None | PT |
| 28 | 31 | F | 25 | − | + | IVIg, rhTPO | GH, PT |
| 29 | 52 | M | 15 | + | − | Steroid, RTX, rhTPO | EC, PT |
| 30 | 28 | F | 21 | + | + | Steroid, rhTPO | GUH, EC |
| 31 | 33 | M | 9 | + | − | Steroid, RTX, CsA | PT |
| 32 | 56 | F | 11 | − | + | Steroid, IVIg | EC, EP, PT |
| Patient no. | Age, y | Sex | Platelet count, ×109/L | Antiplatelet antibody target | Major previous drugs | Bleeding symptoms | |
|---|---|---|---|---|---|---|---|
| GPIIb/IIIa | GPIb/IX | ||||||
| 1 | 41 | F | 12 | + | − | Steroid, VCR | EC, EP |
| 2 | 46 | M | 18 | + | − | Steroid, IVIg, eltrombopag | EC |
| 3 | 22 | F | 6 | − | + | Steroid, RTX | EC, GH |
| 4 | 37 | F | 25 | + | − | Steroid, rhTPO | EC, PT |
| 5 | 28 | M | 22 | + | − | Steroid, rhTPO, IVIg | EC, PT |
| 6 | 33 | M | 5 | + | + | Steroid, rhTPO, RTX | EC, GH |
| 7 | 45 | F | 32 | − | − | Steroid, IVIg, rhTPO, RTX | PT |
| 8 | 52 | F | 14 | − | + | Steroid, VCR | PT, EC |
| 9 | 35 | F | 20 | + | − | Steroid, RTX, eltrombopag | EC |
| 10 | 60 | M | 31 | + | − | Steroid, rhTPO | GH |
| 11 | 40 | F | 19 | − | − | IVIg, steroid | EC |
| 12 | 23 | M | 17 | + | − | Steroid, RTX | EC |
| 13 | 65 | F | 21 | − | + | IVIg, steroid, rhTPO | EP |
| 14 | 47 | M | 16 | + | − | Steroid, RTX, danazol | GH, EP |
| 15 | 29 | F | 8 | + | + | Steroid, RTX | GUH, EP |
| 16 | 25 | M | 23 | − | − | Steroid, IVIg, rhTPO, RTX | PT |
| 17 | 32 | M | 3 | + | + | Steroid, IVIg, rhTPO, RTX | EC, PT, GH |
| 18 | 42 | F | 15 | − | + | Steroid, danazol, RTX | PT |
| 19 | 38 | F | 8 | + | − | Steroid, splenectomy, RTX | EP |
| 20 | 50 | M | 10 | + | − | IVIg, rhTPO, steroid | PT |
| 21 | 57 | M | 7 | + | − | Steroid, thalidomide, CsA | EC, PT |
| 22 | 40 | F | 11 | − | + | Steroid, VCR | EC, GH |
| 23 | 72 | F | 23 | + | − | Steroid | GH |
| 24 | 45 | M | 36 | − | − | Steroid, RTX | EC |
| 25 | 59 | F | 12 | − | + | None | EC, PT |
| 26 | 48 | F | 19 | + | − | Steroid, RTX, rhTPO | EP |
| 27 | 55 | M | 22 | − | − | None | PT |
| 28 | 31 | F | 25 | − | + | IVIg, rhTPO | GH, PT |
| 29 | 52 | M | 15 | + | − | Steroid, RTX, rhTPO | EC, PT |
| 30 | 28 | F | 21 | + | + | Steroid, rhTPO | GUH, EC |
| 31 | 33 | M | 9 | + | − | Steroid, RTX, CsA | PT |
| 32 | 56 | F | 11 | − | + | Steroid, IVIg | EC, EP, PT |
+ indicates antiplatelet antibody positive; − indicates antiplatelet antibody negative.
CsA, cyclosporin; EC, ecchymoses; EP, epistaxis; F, female; GH, gingival hemorrhage; GP, glycoprotein; GUH, genitourinary hemorrhage; IVIg, IV γ-globulin; M, male; PT, petechiae; rhTPO, recombinant human thrombopoietin; RTX, rituximab; VCR, vincristine.
Chidamide
Chidamide (CSO55) was provided by Shenzhen ChipScreen Biosciences (Shenzhen, China) dissolved in water. Chidamide solution was prepared fresh for each use.
Antibodies and flow cytometry
Mouse
Phycoerythrin (PE)-Cy5–labeled rat anti-mouse CD3e, allophycocyanin-labeled rat anti-mouse CD25, fluorescein isothiocyanate (FITC)-labeled rat anti-mouse CD4, PE/PE-Cy5–labeled rat anti-mouse Foxp3, and PE-labeled rat anti-mouse CTLA4 (CD152) were purchased from eBioscience (San Diego, CA).
Human
PE/FITC-labeled mouse anti-human CD25, PE-Cy5/FITC–labeled mouse anti-human CD4, allophycocyanin-labeled mouse anti-human Foxp3, and PE-labeled mouse anti-human CTLA4 (CD152) were purchased from eBioscience or BD Biosciences. Recombinant human IL-2 was purchased from Chiron. Clinical-grade beads coated with anti-CD3 and anti-CD28 antibodies (T-cell expander) were purchased from Dynal Biotech.
Animals
Passive ITP model
Male C57BL/6J mice (6-8 weeks old; platelet count range, 880-1240 × 109/L; median platelet count, 1045 × 109/L) were injected IV with anti-platelet monoclonal antibody (mAb; rat anti-mouse CD41, clone MWReg30; BD Biosciences) at an initial dose of 0.3 mg/kg body weight and follow-up doses of 0.1 mg/kg every 36 hours. To test whether low-dose chidamide could repair thrombocytopenia, we intragastrically administered chidamide (0, 0.01, 0.1, 0.5, and 5 mg/kg) to passive ITP mice simultaneously with the first IV injection of anti-platelet mAb.
To analyze platelet count, whole-blood samples (5 μL) were collected from the vein of lower extremities and mixed with anticoagulant ACD solution (38 mM citric acid, 75 mM sodium citrate, 100 mM dextrose). We counted peripheral blood platelets with a hematology analyzer (KX-21N; Sysmex) 120 hours after the first administration of anti-CD41 mAb. The optimal mAb dosage was determined according to the platelet count. Then platelet counts were measured at 0, 24, 48, 72, and 120 hours after the first anti-CD41 treatment. The time for platelet recovery in the passive ITP mice model was 5 to 8 days without treatment.8,9
Active ITP model
Male C57BL/6J mice that were used as platelet donors were purchased from the Center for New Drug Evaluation of Shandong University. Severe combined immunodeficient mice on a C57BL/6 background (J001913, 6-8 weeks old; platelet count range, 840-1310 × 109/L; median platelet count, 1120 × 109/L), which were used as spleen cell transfer recipients and chidamide recipients, were purchased from The Jackson Laboratory (Bar Harbor, ME). C57BL/6 CD61-knockout mice (B6.129S2-Itgb3tm1Hyn/JSemJ; stock no. 008819) were provided by Junling Liu (Shanghai Jiaotong University School of Basic Medicine).
We immunized CD61-knockout mice against CD61+ platelets (also called Wistar platelet-immunized mice). Then, 5 × 104 splenocytes from these mice were engrafted into CD61+ severe combined immunodeficient mice to establish active ITP mice, which exhibited profound thrombocytopenia. Bleeding-related mortality occurred in some mice within 28 days of transfer. Chidamide (0.1 mg/kg) was administered intragastrically simultaneously with the splenocyte transfer and 3 times per week after splenocyte transfer. The time for platelet recovery in the active ITP mice model was ∼28 to 35 days without treatment.10
Animal studies were approved by the Animal Care and Use Committee of Qilu Hospital and were conducted under the guidelines for animal care and use of Shandong University.
Splenocyte preparation
After receiving chidamide treatment for 4 weeks for the active ITP model, mice were euthanized, and spleens were harvested. Single-cell suspensions were prepared. Cells were treated with hemolysis buffer to remove red blood cells. After washing in phosphate buffered saline, we counted cells and split them for flow cytometry staining.
Immunofluorescence and flow cytometry
To stain intracellular Foxp3, cells were incubated with anti-CD4 and anti-CD25 mAbs (30 minutes, 4°C, dark), as described in supplemental Methods (available on the Blood Web site). Cells were then washed, fixed, and stained with anti-Foxp3 mAb. To stain membrane CTLA4 (CD152), cells were incubated with anti-CD4 and anti-CD152 mAbs. To stain intracellular CTLA4 (CD152) in Treg cells, we incubated them with anti-CD4 and anti-CD25 mAbs. Cells were then washed, fixed, permeabilized, and stained with anti-CTLA4 and anti-Foxp3 mAbs.11
Isolation, 5-chloromethylfluorescein diacetate labeling, opsonization of platelets, and evaluation of phagocytic capacity of monocyte-derived macrophages
Preparation, culture and analysis of ITP patient PBMCs
Whole blood was obtained from ITP patients and healthy volunteers by venipuncture into EDTA. PBMCs were separated by density gradient centrifugation using Ficoll-Hypaque (1.077 g/mL). Isolated PBMCs (1 × 106) were cultured in complete RPMI 1640 medium, with or without 10 nM chidamide. Cells were stimulated with phytohemagglutinin (5 µg/mL; Sigma) and recombinant interleukin-2 (IL-2; 5 ng/mL; PeproTech) and incubated for 72 hours (37°C, 5% CO2).14 After incubation, cells were collected and the percentage of CD4+CD25+Foxp3+ Treg cells was analyzed by flow cytometry, as described above.
Purification of human CD4+CD25+ and CD4+CD25− T cells
CD4+CD25+ Treg cell populations were isolated from human PBMCs and enriched using a CD4+CD25+ Regulatory T Cell Isolation Kit with a MidiMACS Separator, according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). The purity was >95% for the CD4+CD25+ Treg cells and >98% for the CD4+CD25− T cells, as measured by flow cytometry. Purified cells were suspended in RPMI 1640 medium.
Immunosuppression assays of CD4+CD25+ T cells
CD4+CD25+ Treg cells were isolated from human PBMCs, as described above. Isolated naive human CD4+CD25− effector T cells (4 × 106 cells per milliliter) with 5,6-carboxyfluorescein diacetate succinimidyl ester (2.5 µM, 37°C, 30 minutes) were added to 96-well plates and stimulated with anti-CD3 and anti-CD28–coated beads from the Treg Suppression Inspector kit (Miltenyi Biotec), in the presence or absence of ITP patient Treg cells. CD4+CD25+ Treg cells and CD4+CD25− effector T cells were added in a 1:4 ratio. Cells were cultured or not with 10 nM chidamide (37°C, 5% CO2). On day 5, the proliferation of effector cells was determined by flow cytometry.
Chromatin immunoprecipitation assay
PBMCs were isolated from the peripheral blood of ITP patients and healthy controls and prepared for chromatin immunoprecipitation (ChIP). ChIP assays use highly specific antibodies to immunoprecipitate specific protein/DNA complexes.15 The antibody used was a ChIP-grade anti-histone H3 (acetyl K27) antibody (ab4729; Abcam). We then used real-time quantitative PCR (qPCR) to detect the human CTLA4 gene in the purified DNA (primers detailed in Table 2).
Gene-specific primers for qPCR of human CTLA4 Ig gene in ChIP-Seq
| Primers | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| 1 | TGGAGGATGAGAAGGAGTAT | AGCATTCCCTCCCATT |
| 2 | AATGGGAGGGAATGCTG | CCTACTCCAGTCCGTCCA |
| 3 | TGTTAGATCATTGGTCCTGTC | AAGAGATTAGAGCCGTCCA |
| 4 | TCAAGGGACCATTAGAAGG | TGGGCTAATGGCAGGAT |
| 5 | TAAACCCACGGCTTCCT | GCTTTGATCCCAGATATGTATT |
| Primers | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| 1 | TGGAGGATGAGAAGGAGTAT | AGCATTCCCTCCCATT |
| 2 | AATGGGAGGGAATGCTG | CCTACTCCAGTCCGTCCA |
| 3 | TGTTAGATCATTGGTCCTGTC | AAGAGATTAGAGCCGTCCA |
| 4 | TCAAGGGACCATTAGAAGG | TGGGCTAATGGCAGGAT |
| 5 | TAAACCCACGGCTTCCT | GCTTTGATCCCAGATATGTATT |
Six ITP patients (3 females, 3 males; age range, 32-57 years; median age, 42 years; platelet count range, 3-18 × 109/L; median platelet count, 10 × 109/L) and 4 healthy controls (2 females, 2 males) were screened using the ChIP assay to detect the CTLA4 gene histone acetylation level in peripheral lymphocytes. PBMCs were cultured as above, with or without 10 nM chidamide. Cells were stimulated with phytohemagglutinin (5 µg/mL; Sigma) and recombinant IL-2 (5 ng/mL, PeproTech) for 72 h (37°C, 5% CO2). After incubation, cells were collected for the ChIP assay.
The ChIP assay was performed as described in supplemental Methods. The efficiency (ChIP/total input) of gene immune coprecipitation was calculated as follows: % (ChIP/total input) = 2[Ct (Input) − Ct (ChIP)] × 100. Relative expression of the CTLA4 gene can reflect the acetylation level of the histone H3K27 on the CTLA4 gene.
Quantitative real-time PCR
Total RNA was extracted from the spleens of active ITP mice. qPCR was performed as described in supplemental Methods. The primer sequences used for qPCR of the mouse CTLA4 gene are detailed in Table 3.
Lentivirus interference of CTLA4 gene expression
Briefly, murine splenocytes were transduced with lentivirus encoding the murine CTLA4 short hairpin RNA (shRNA; target sequence: GGACGCAGATTTATGTCAT). To select transduced cells, we added puromycin to the culture medium (1 mg/mL) and incubated it for 48 hours. CTLA4 downregulation was measured by qPCR. Then, murine CTLA4 shRNA–transfected splenocytes were injected into mice via the caudal vein to interfere with CTLA4 expression.
Statistical analysis
All data were analyzed by SPSS software and are presented as mean ± standard deviation. The Student t test was used to compare means; P < .05 was considered statistically significant.
Results
Low-dose chidamide ameliorated transient thrombocytopenia in the passive ITP model
We found that administration of low-dose chidamide (0.1 mg/kg) twice a week significantly improved platelet counts compared with other dosages in passive ITP mice 120 hours after immunization (1186 ± 83.04 × 109/L at 0.1 mg/kg vs 447 ± 61.92 × 109/L at 0 mg/kg, 730.6 ± 67.73 × 109/L at 0.01 mg/kg, 749.2 ± 80.09 × 109/L at 0.5 mg/kg, and 437.4 ± 44.09 × 109/L at 5 mg/kg, respectively; n = 5; P < .01; Figure 1A).

Low-dose chidamide reversed thrombocytopenia in passive ITP mice. (A) Low-dose chidamide (0.1 mg/kg, twice a week) improved platelet counts in mice immunized with monoclonal rat anti-mouse CD41 platelet antibody at 120 hours compared with the other dosages. (B) Low-dose chidamide (0.1 mg/kg, twice a week) improved transient thrombocytopenia in passive ITP mice at 72 and 120 hours after immunization. *P < .05, **P < .01.
Low-dose chidamide reversed thrombocytopenia in passive ITP mice. (A) Low-dose chidamide (0.1 mg/kg, twice a week) improved platelet counts in mice immunized with monoclonal rat anti-mouse CD41 platelet antibody at 120 hours compared with the other dosages. (B) Low-dose chidamide (0.1 mg/kg, twice a week) improved transient thrombocytopenia in passive ITP mice at 72 and 120 hours after immunization. *P < .05, **P < .01.
We then treated passive ITP mice with 0.1 mg/kg chidamide and counted platelets 24, 72, and 120 hours after the first administration of anti-CD41 mAb. Low-dose chidamide significantly increased the platelet count in ITP mice at 72 hours (365 ± 38.62 × 109/L vs 160 ± 45.28 × 109/L; n = 4; P < .05) and at 120 hours (1655 ± 167.9 × 109/L vs 740 ± 168.2 × 109/L; n = 4; P < .05; Fig 1B) after immunization compared with untreated ITP mice, suggesting that chidamide had therapeutic effects on ITP.
Low-dose chidamide ameliorated transient thrombocytopenia in the active ITP model
To study the therapeutic effect of low-dose chidamide further, we used an active murine model of ITP. One week after splenocyte transfer, the platelet count decreased significantly. The chidamide-treated group had higher platelet counts but no significant difference on day 7 (319.2 ± 47.4 × 109/L vs 244 ± 66.5 × 109/L; chidamide, n = 12; control, n = 10; P > .05). After splenocyte transfer, the chidamide-treated mice had significantly higher platelet counts compared with controls at day 14 (166.4 ± 29.7 × 109/L vs 76.25 ± 30.88 × 109/L; chidamide, n = 11; control, n = 8; P < .05) and at day 21 (319.1 ± 61.71 × 109/L vs 147.1 ± 56.64 × 109/L; chidamide, n = 11; control n = 7; P < .05; Figure 2A).
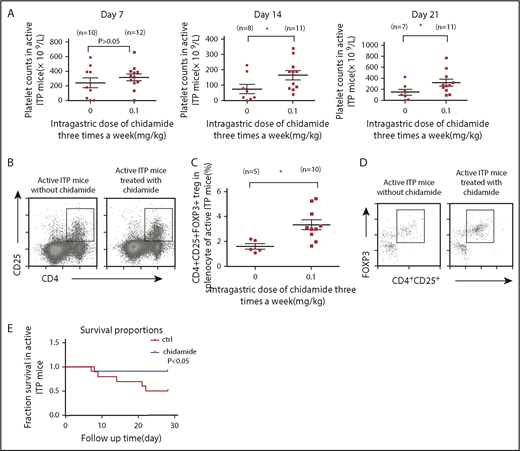
Low-dose chidamide reversed thrombocytopenia, enhanced CD4+CD25+Foxp3+ Treg cells in splenocytes, and decreased mortality from bleeding in active ITP mice. (A) Low-dose chidamide (0.1 mg/kg, 3 times per week) ameliorated transient thrombocytopenia in mice immunized by Wistar platelets on days 14 and 21. (B) Flow cytometry revealed that low-dose chidamide enhanced the expression of CD4+CD25+ Treg cells in active ITP mice. (C-D) Low-dose chidamide enhanced the percentage and number of CD4+CD25+Foxp3+ Treg cells in active ITP mice. (E) Low-dose chidamide (0.1 mg/kg) 3 times per week decreased bleeding mortality caused by severe thrombocytopenia and prolonged survival in active ITP mice. *P < .05. ctrl, control.
Low-dose chidamide reversed thrombocytopenia, enhanced CD4+CD25+Foxp3+ Treg cells in splenocytes, and decreased mortality from bleeding in active ITP mice. (A) Low-dose chidamide (0.1 mg/kg, 3 times per week) ameliorated transient thrombocytopenia in mice immunized by Wistar platelets on days 14 and 21. (B) Flow cytometry revealed that low-dose chidamide enhanced the expression of CD4+CD25+ Treg cells in active ITP mice. (C-D) Low-dose chidamide enhanced the percentage and number of CD4+CD25+Foxp3+ Treg cells in active ITP mice. (E) Low-dose chidamide (0.1 mg/kg) 3 times per week decreased bleeding mortality caused by severe thrombocytopenia and prolonged survival in active ITP mice. *P < .05. ctrl, control.
Increased percentage and number of CD4+CD25+Foxp3+ Treg cells in splenocytes of active ITP mice
After 4 weeks of low-dose chidamide, the percentage of CD4+CD25+ Treg cells was increased significantly in chidamide-treated ITP mice compared with untreated ITP mice (8.255% ± 2.421% vs 3.582% ± 0.508%; chidamide, n = 10; control, n = 5; P < .05; Figure 2B). Consistent with these findings, CD4+CD25+Foxp3+ Treg cells were increased significantly in low-dose chidamide-treated ITP mice (3.346% ± 0.398% vs 1.989% ± 0.301%; chidamide, n = 10; control, n = 5; P < .05; Figure 2C-D). Mortality caused by severe thrombocytopenia was reduced significantly in the chidamide-treated mice (8.3% vs 50%; P < .05; Figure 2E).
Low-dose chidamide decreased monocyte/macrophage phagocytic capacity
Macrophages derived from ITP patient monocytes and treated with low-dose chidamide (10 nM) demonstrated less phagocytosis than did those without chidamide treatment (mean phagocytic index: 1.575 ± 0.317 vs 5.508 ± 0.414; n = 4; P < .01; Figure 3). Low-dose chidamide treatment significantly decreased the phagocytic capacity of monocyte-derived macrophages.
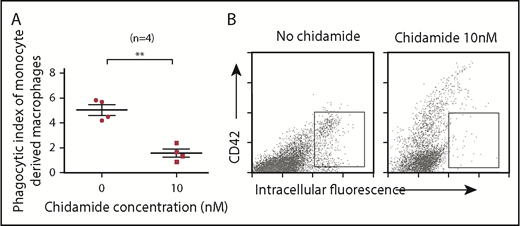
Low-dose chidamide inhibited macrophage phagocytosis of antibody-coated platelets in vitro. (A) Low-dose chidamide (10 nM) inhibited macrophage phagocytosis of mouse anti-human antibody-coated platelets in vitro (n = 4). (B) Intracellular fluorescence–positive scatters indicates platelets phagocytosed by macrophages. CD42+ scatter indicates adhered, but unphagocytosed, platelets. **P < .01.
Low-dose chidamide inhibited macrophage phagocytosis of antibody-coated platelets in vitro. (A) Low-dose chidamide (10 nM) inhibited macrophage phagocytosis of mouse anti-human antibody-coated platelets in vitro (n = 4). (B) Intracellular fluorescence–positive scatters indicates platelets phagocytosed by macrophages. CD42+ scatter indicates adhered, but unphagocytosed, platelets. **P < .01.
Increased CD4+CD25+Foxp3+ Treg cells in PBMCs from ITP patients
To study the effect of low-dose chidamide on ITP patients’ Treg cells, we isolated PBMCs from ITP patients and healthy volunteers and cultured them with or not with low-dose chidamide (0, 1, 10 nM). After 72 hours in culture with 10 nM chidamide, the percentage of CD4+CD25+Foxp3+ Treg cells from ITP patients was increased significantly compared with other concentrations of chidamide (2.441% ± 0.5434% at 10 nM vs 1.686% ± 0.4341% at 1 nM and 1.413% ± 0.4504% at 0 nM, n = 8; P < .05; Figure 4A). The percentage of CD4+CD25+Foxp3+ Treg cells in healthy volunteers did not change significantly after culture with chidamide (9.144% ± 2.931% at 10 nM vs 10.05% ± 2.321% at 1 nM and 7.714% ± 2.204% at 0 nM; n = 8; P > .05; Figure 4B). The percentage of CD4+ T cells did not change significantly after culture with chidamide (71.07 ± 1.73% at 10 nM vs 62.15% ± 3.671% at 1 nM and 68.13% ± 2.71% at 0 nM; n = 8; P > .05; Figure 4C). These results indicate that low-dose chidamide increased the number and percentage of CD4+CD25+Foxp3+ Treg cells in vitro in ITP patients.
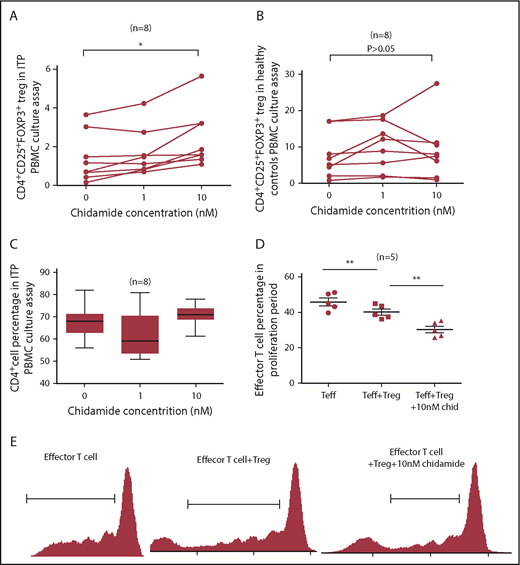
Low-dose chidamide increased CD4+CD25+Foxp3+ Treg cells in ITP patient PBMCs and enhanced immunosuppression function of CD4+CD25+ Treg cells. (A) Low-dose chidamide (10 nM) increased the percentage and number of CD4+CD25+Foxp3+ Treg cells in ITP patients. (B) We found no significant change in the percentages of CD4+CD25+Foxp3+ Treg cells in healthy volunteers after chidamide treatment. (C) The percentage of CD4+ T cells did not vary significantly before and after chidamide treatment. (D-E) Low-dose chidamide (10 nM) stimulated Treg cells to inhibit effector T cells. Coculture of chidamide and Treg cells resulted in a significantly lower proportion of effector T cells during the proliferation stage than those without chidamide treatment. *P < .05, **P < .01.
Low-dose chidamide increased CD4+CD25+Foxp3+ Treg cells in ITP patient PBMCs and enhanced immunosuppression function of CD4+CD25+ Treg cells. (A) Low-dose chidamide (10 nM) increased the percentage and number of CD4+CD25+Foxp3+ Treg cells in ITP patients. (B) We found no significant change in the percentages of CD4+CD25+Foxp3+ Treg cells in healthy volunteers after chidamide treatment. (C) The percentage of CD4+ T cells did not vary significantly before and after chidamide treatment. (D-E) Low-dose chidamide (10 nM) stimulated Treg cells to inhibit effector T cells. Coculture of chidamide and Treg cells resulted in a significantly lower proportion of effector T cells during the proliferation stage than those without chidamide treatment. *P < .05, **P < .01.
Enhanced immunosuppressive function of ITP patient CD4+CD25+ Treg cells in vitro
We cultured CD4+CD25+ Treg cells and CD4+CD25− effector T cells at a 1:4 ratio, with or without 10 nM chidamide, and detected proliferation of effector T cells using 5,6-carboxyfluorescein diacetate succinimidyl ester. The percentage and number of effector T cells in the proliferation phase were significantly decreased after co-incubation with CD4+CD25+ Treg cells. Low-dose chidamide prompted Treg cells to inhibit proliferation of effector T cells (30.26% ± 1.885% at Treg + 10 nM chidamide vs 40.21% ± 1.731% at Treg and 45.9% ± 2.181% at control; n = 5; P < .01; Figure 4D-E), suggesting that the immunosuppressive function of CD4+CD25+ Treg cells was enhanced in vitro.
Increased surface expression of CTLA4 in splenocytes of active ITP mice
Recent research suggested that CTLA4 levels might be associated with the pathogenesis of acute ITP.16 To understand the immunological mechanism of low-dose chidamide, we measured surface CD4+CTLA4+ T cells in splenocytes from active ITP mice. After 4 weeks of treatment, the percentage of CD4+CTLA4+ T cells was increased significantly in active ITP mice (4.938% ± 0.65% vs 2.25% ± 0.80%; P < .05; Figure 5A). These results suggested that the surface phenotypes of T cells were altered and higher expression of CTLA4 was acquired after low-dose chidamide treatment.
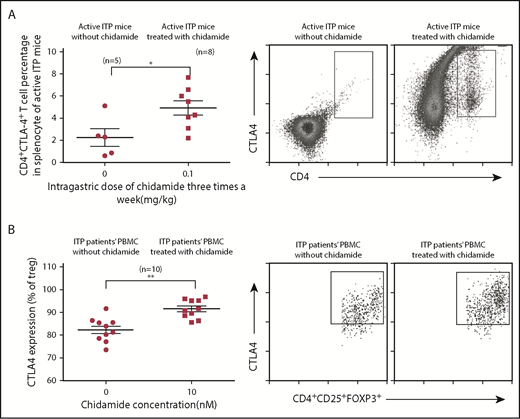
Low-dose chidamide enhanced CTLA4 expression in splenocytes from active ITP mice and in CD4+CD25+Foxp3+ Treg cells from PBMCs from ITP patients. (A) Low-dose chidamide enhanced CD4+CTLA4+ T cell numbers and percentages in splenocytes from active ITP mice. (B) Low-dose chidamide enhanced CTLA4 expression in CD4+CD25+FOXP3+ Treg cells in PBMCs from ITP patients. *P < .05, **P < .01.
Low-dose chidamide enhanced CTLA4 expression in splenocytes from active ITP mice and in CD4+CD25+Foxp3+ Treg cells from PBMCs from ITP patients. (A) Low-dose chidamide enhanced CD4+CTLA4+ T cell numbers and percentages in splenocytes from active ITP mice. (B) Low-dose chidamide enhanced CTLA4 expression in CD4+CD25+FOXP3+ Treg cells in PBMCs from ITP patients. *P < .05, **P < .01.
Enhanced CD4+CD25+Foxp3+ Treg cells expressing intracellular CTLA4 in ITP patient PBMCs
We found that low-dose chidamide treatment (10 nM) enhanced CD4+CD25+Foxp3+ Treg cells expressing intracellular CTLA4 in the PBMC culture assay (91.56% ± 1.28% vs 82.33% ± 1.66%; n = 10; P < .01; Figure 5B).
Correction of reduced acetylation of CTLA4 in ITP patients
We performed a ChIP-Seq assay to determine the histone H3K27 acetylation levels in CTLA4 genes from ITP patients and healthy controls. The nuclei were extracted from the formaldehyde-fixed cells, and chromatin was broken into 200- to 500–bp fragments with ultrasonic agarose gel to detect DNA fragment sizes (Figure 6A). Primers 1 through 5 were designed to target the human CTLA4 genome (Figure 6B). We found that the relative expression was significantly lower in ITP patients than in controls (primer 1: 0.08638 ± 0.02326 vs 0.1943 ± 0.01905, P < .05; primer 2: 0.05112 ± 0.01493 vs 0.1398 ± 0.02233, P < .01; primer 3: 0.09305 ± 0.02699 vs 0.2658 ± 0.03640, P < .05; primer 4: 0.07633 ± 0.02408 vs 0.3073 ± 0.07249, P < .01; primer 5: 0.08392 ± 0.01907 vs 0.203 ± 0.01453, P < .01; ITP, n = 6; control, n = 4; Figure 6C), suggesting that acetylation of histone H3K27 on the CTLA4 gene in ITP patients was significantly lower than in healthy controls. We hypothesized that reduced acetylation of histone H3K27 was associated with decreased CTLA4 expression and impaired immunological tolerance and, therefore, contributed to the pathology of ITP.
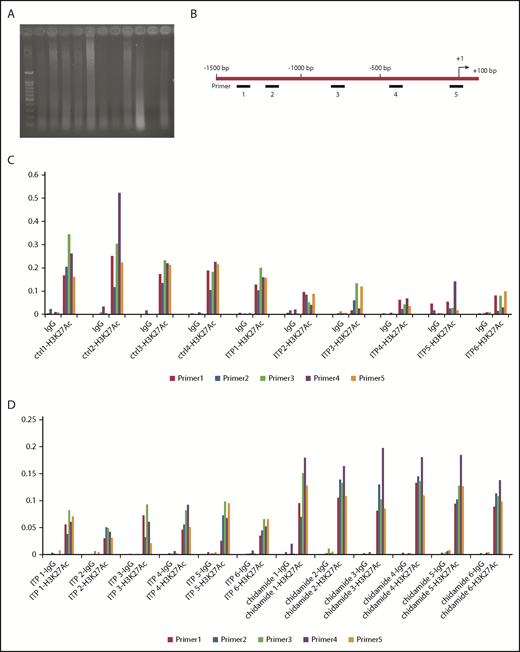
ITP patients had less histone acetylation within the CTLA4 gene. ChIP-Seq used the H3K27-specific antibodies to capture DNA fragments of the CTLA4 gene in human PBMCs and then used qPCR to amplify the captured fragments. Relative expression of the CTLA4 gene can reflect the acetylation level of histone H3K27. (A) DNA ultrasonic broken image. Healthy controls (ctrl1-4), and ITP patients’ (ITP1-6) chromatin were broken into 200- to 500–bp fragments with ultrasonic agarose gel to detect fragment size, and the ultrasonic broken DNA samples were quantified. (B) Location of primers 1 to 5 within the human CTLA4 gene. (C) ITP patients had less acetylated H3K27 expression in the CTLA4 gene compared with that in the control group, as assessed by qPCR. (D) PBMCs were collected for ChIP-Seq to detect the acetylation level of histone H3K27 in the CTLA4 gene. ITP patients treated with chidamide had significantly more acetylated H3K27 than did those without chidamide treatment.
ITP patients had less histone acetylation within the CTLA4 gene. ChIP-Seq used the H3K27-specific antibodies to capture DNA fragments of the CTLA4 gene in human PBMCs and then used qPCR to amplify the captured fragments. Relative expression of the CTLA4 gene can reflect the acetylation level of histone H3K27. (A) DNA ultrasonic broken image. Healthy controls (ctrl1-4), and ITP patients’ (ITP1-6) chromatin were broken into 200- to 500–bp fragments with ultrasonic agarose gel to detect fragment size, and the ultrasonic broken DNA samples were quantified. (B) Location of primers 1 to 5 within the human CTLA4 gene. (C) ITP patients had less acetylated H3K27 expression in the CTLA4 gene compared with that in the control group, as assessed by qPCR. (D) PBMCs were collected for ChIP-Seq to detect the acetylation level of histone H3K27 in the CTLA4 gene. ITP patients treated with chidamide had significantly more acetylated H3K27 than did those without chidamide treatment.
PBMCs were then divided into 2 sets. One set was cultured with 10 nM chidamide, and the other was not. A ChIP assay was performed to detect histone H3K27 on the CTLA4 gene. Treated cells showed significantly higher acetylated H3K27 on the CTLA4 gene compared with cells that did not receive chidamide treatment by qPCR (primer 1: 0.1001 ± 0.0075 vs 0.0449 ± 0.0072, P < .01; primer 2: 0.1172 ± 0.0112 vs 0.04975 ± 0.00576, P < .01; primer 3: 0.1268 ± 0.00745 vs 0.07895 ± 0.00735, P < .05; primer 4: 0.1744 ± 0.00845 vs 0.063 ± 0.00668, P < .05; primer 5: 0.1099 ± 0.00675 vs 0.05637 ± 0.01107, P < .01; n = 6; Figure 6D).
Increased CTLA4 mRNA expression in active ITP mice
We next administered chidamide to active ITP mouse models and measured CTLA4 gene expression by qPCR. Low-dose chidamide (3 times per week) significantly increased relative CTLA4 mRNA expression in ITP mice immunized by Wistar platelets (active model, 0.05181 ± 0.014 vs 0.03321 ± 0.01287; control, n = 5; chidamide, n = 11; P < .05; Figure 7A).
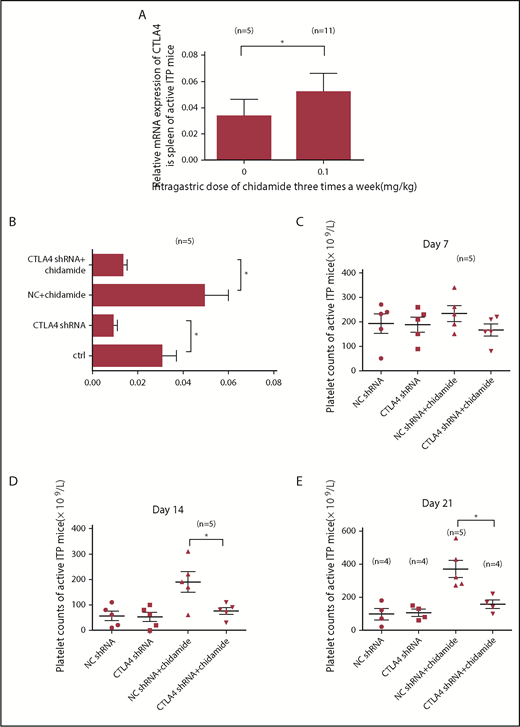
Low-dose chidamide increased CTLA4 mRNA expression, and CTLA4 shRNA interference prevented the platelet count from improving with chidamide treatment. (A) Low-dose chidamide increased relative mRNA expression of the CTLA4 gene in active ITP mice. (B) CTLA4 shRNA interference decreased CTLA4 gene expression in the CTLA4 shRNA group and in the CTLA4 shRNA + chidamide group in the active ITP mice. (C-E) CTLA4 shRNA transference offset the low-dose chidamide treatment in active ITP mice on days 14 and 21. *P < .05.
Low-dose chidamide increased CTLA4 mRNA expression, and CTLA4 shRNA interference prevented the platelet count from improving with chidamide treatment. (A) Low-dose chidamide increased relative mRNA expression of the CTLA4 gene in active ITP mice. (B) CTLA4 shRNA interference decreased CTLA4 gene expression in the CTLA4 shRNA group and in the CTLA4 shRNA + chidamide group in the active ITP mice. (C-E) CTLA4 shRNA transference offset the low-dose chidamide treatment in active ITP mice on days 14 and 21. *P < .05.
CTLA4 shRNA interference offset the low-dose chidamide treatment in active ITP mice
To determine whether the mechanism of action of low-dose chidamide was via the CTLA4 gene, we performed a lentivirus interference test. We blocked CTLA4 gene expression in active ITP mice. Mice were divided into 4 groups: control lentivirus (NC), CTLA4 lentivirus (CTLA4 shRNA), NC + chidamide, and CTLA4 shRNA + chidamide. shRNA transfection successfully silenced the CTLA4 gene; low-dose chidamide–treated ITP mice transfected with CTLA4 shRNA displayed significantly reduced CTLA4 mRNA expression compared with those transfected with the control lentivirus (0.008954 ± 0.002 vs 0.03041 ± 0.0064; n = 5; P < .05; Figure 7B).
Platelet counts in chidamide-treated ITP mice after CTLA4 shRNA transfection were not significantly different from those in mice who received control lentivirus on day 7 (166 ± 24.8 × 109/L vs 234 ± 32.34 × 109/L; n = 5; P > .05; Figure 7C), but they were decreased significantly compared with control mice on day 14 (76 ± 13.3 × 109/L vs 190 ± 40.37 × 109/L; n = 5; P < .05; Figure 7D) and on day 21 (157.5 ± 25.29 × 109/L vs 370 ± 53.2 × 109/L; NC + chidamide, n = 5; the other groups, n = 4; P < .05; Figure 7E). Reduced platelet destruction was offset by excluding the role of CTLA4.
Discussion
Although current therapeutic regimens have replaced the generalized immunosuppressive strategies in the treatment of autoimmune diseases, a substantial proportion of ITP patients fail to achieve clinical remission and to reestablish tolerance. Further study of the pathogenesis of ITP is urgently needed to improve patient care.
Antibody-mediated platelet damage by macrophages was the main mechanism of action of the passive ITP model.17-19 Our data showed that low-dose chidamide significantly reduced phagocytosis of antibody-coated platelets by monocyte-derived macrophages from ITP patients in vitro and alleviated thrombocytopenia in the passive ITP model. We propose that chidamide’s main mechanism of action might be through direct inhibition of macrophage phagocytosis in the passive ITP model. Previous reports showed that HDACi’s decreased macrophage phagocytosis and killing of Escherichia coli and Staphylococcus aureus by downregulating the expression of phagocytic receptors and impairing expression of nicotinamide adenine dinucleotide phosphate oxidase subunits and inducible nitric oxide synthase.20,21 Whether these mechanisms play a role in chidamide’s effect on passive ITP mice requires further investigation.
Naturally occurring CD4+CD25+Foxp3+ Treg cells are essential for maintaining immunological self-tolerance and immune homeostasis.22,23 Treg cells potently suppress the proliferation and cytokine production of effector CD4+ and CD8+ T cells by inhibiting IL-2 gene transcription.24-27 Treg cells also inhibit B cell proliferation and antibody production,28 suppress proinflammatory activation of macrophages,29 and modulate monocyte-to-macrophage differentiation.30,31 Decreased numbers and impaired functions of Treg cells have been reported in patients with various autoimmune diseases, including ITP.1,32-35 In the active ITP model, low-dose chidamide significantly enhanced natural CD4+CD25+Foxp3+ T cells in splenocytes. Low-dose chidamide significantly increased platelet counts and decreased mortality caused by fatal thrombocytopenia in the active ITP mice. A higher frequency of CD4+CD25+Foxp3+ T cells and increased immunosuppressive functions of CD4+CD25+ Treg cells were observed when ITP patient PBMCs were cultured with low-dose chidamide. Chidamide may increase platelet counts and decrease mortality in the active ITP model through enhancing the number and function of naturally occurring Treg cells.
CTLA4 blocks the interaction of B7 proteins with CD28 and, therefore, prevents initial T cell activation. CTLA4 is highly expressed on T cells and inhibits effector T cells through regulatory T cells and tumor growth factor-β.36,37 CTLA4 expression was associated with a higher incidence of CD25hiFoxp3hi Treg cells.38 Dysregulated CTLA4 expression leads to many autoimmune diseases, including ITP.39 In this study, low-dose chidamide significantly increased CTLA4 mRNA expression and promoted CD4+CTLA4+ T cell percentages in splenocytes from active ITP mice. Higher intracellular CTLA4 expression was also observed in CD4+CD25+Foxp3+ Treg cells in vitro after chidamide treatment. CTLA4 shRNA interference neutralized the therapeutic effect of low-dose chidamide in ITP. Our findings indicated that low-dose chidamide promoted Treg cell proliferation through upregulation of surface CTLA4 expression on T lymphocytes and intracellular CTLA4 expression on Treg cells.
Epigenetic abnormalities, mechanisms closely related to altered gene expression, are involved in T cell differentiation and functional development in autoimmune diseases.40-43 Aberrant histone methylation is involved in the pathogenesis of ITP.44 In the present study, we performed a ChIP-Seq assay and found that ITP patients had lower histone acetylation within the CTLA4 gene compared with healthy controls, which led to reduced CTLA4 expression. We propose that a histone acetylation abnormality of H3K27 in the CTLA4 gene in PBMCs leads to dysregulation of CTLA4 protein expression and, therefore, contributes to impaired numbers and functions of Treg cells and pathogenesis of ITP. Our finding that low-dose chidamide increased CD4+CD25+Foxp3+ Treg cells only in ITP patients suggests that ITP may be associated with aberrant histone acetylation of H3K27 in the CTLA4 gene. At doses required for direct antitumor effects, HDACi’s induced profound thrombocytopenia45-47 by inhibiting colony-forming unit–megakaryocyte growth, megakaryocyte proliferation and differentiation, and proplatelet formation.48,49 At much lower concentrations, HDACi’s demonstrated immunomodulatory effects.50 Our study demonstrated that low-dose chidamide restores the histone H3K27 acetylation level in PBMCs isolated from ITP patients and improves CTLA4 mRNA expression in ITP.
In summary, our findings revealed that low-dose chidamide inhibited macrophage phagocytosis, upregulated CTLA4 expression, restored the immunosuppressive effect of natural CD4+CD25+Foxp3+ Treg cells, and alleviated thrombocytopenia in the passive and active ITP models. Importantly, our study is the first to identify that histone acetylation abnormalities of H3K27 in the CTLA4 gene in PBMCs was involved in the pathogenesis of ITP. In conclusion, low-dose chidamide has potential as a novel therapeutic for ITP patients, through modulation of macrophage phagocytosis, natural Treg cells, and CTLA4 gene expression.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Alexandra Helen Marshall (Marshall Medical Communications) for editing the manuscript.
This work was supported by the National Natural Science Foundation of China (grants 81770114, 81770133, 81600091, 81570103, and 81470284), the Major Research Plan of the National Natural Science Foundation of China (grant 91442204), the Major Research Plan of the Natural Science Foundation of Shandong Province (grant ZR2016QZ008), State Key Clinical Specialty of China for Hematological Diseases, and the Taishan Scholar of Shandong Province.
Authorship
Contribution: M.H. designed and funded the study; H. Zhao and Y.M. performed the research; P.L., Y.H., Y.L., P.H., and Y.Z. assisted with the research; H. Zhou, X.L., L.L., and F.J. analyzed the data; H. Zhao wrote the manuscript; M.H., J.P., D.L., and T.S. edited the manuscript; and J.P., H. Zhou, and X.L. helped to fund the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ming Hou, Department of Hematology, Qilu Hospital, Shandong University, 107 West Wenhua Rd, Jinan, Shandong 250012, China; e-mail: houming@medmail.com.cn; and Jun Peng, Shandong Provincial Key Laboratory of Immunohematology, Qilu Hospital, Shandong University, 107 West Wenhua Rd, Jinan, Shandong 250012, China; e-mail: junpeng88@sina.com.cn.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal