

In this issue of Blood, reveal that signaling lymphocyte activation molecule F7 (SLAMF7) may be a potential therapeutic target for modifying the bone marrow (BM) microenvironment in patients with myelofibrosis (MF).1
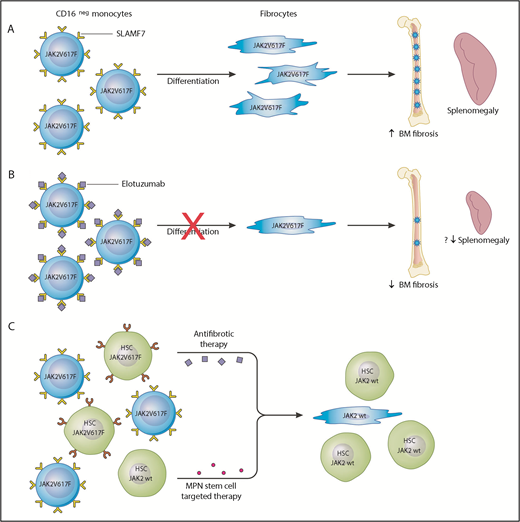
SLAMF7 as a potential therapeutic target in MF. (A) In the peripheral blood of MF patients, there is an increase in the percentage of SLAMF7highCD16– monocytes and this positively correlates with the JAK2V617F allele burden. Clonal involved, monocyte-derived fibrocytes drive BM fibrosis through cytokine production and other unknown mechanisms. They specifically promote deposition of reticulin fibrosis in the BM and may contribute to splenomegaly. (B) The authors showed that monocytes from MF patients treated with the anti-SLAMF7 antibody elotuzumab result in reduction of fibrocyte differentiation. Treatment of a romiplostim-induced myelofibrotic mouse model with elotuzumab resulted in decreased BM fibrosis and reduction in spleen size. This serves as preclinical evidence that elotuzumab may modulate the BM microenvironment in MF patients and provide a more hospitable environment for normal hematopoiesis. (C) Elotuzumab targets the BM microenvironment through inhibiting differentiation of SLAMF7highCD16– to neoplastic fibrocytes; however, it is unlikely that this agent has any effect on the MPN hematopoietic stem cells. A combination therapy approach targeting the MPN hematopoietic stem cells and restoring the BM microenvironment would likely be the most effective strategy to achieve disease course modification.
SLAMF7 as a potential therapeutic target in MF. (A) In the peripheral blood of MF patients, there is an increase in the percentage of SLAMF7highCD16– monocytes and this positively correlates with the JAK2V617F allele burden. Clonal involved, monocyte-derived fibrocytes drive BM fibrosis through cytokine production and other unknown mechanisms. They specifically promote deposition of reticulin fibrosis in the BM and may contribute to splenomegaly. (B) The authors showed that monocytes from MF patients treated with the anti-SLAMF7 antibody elotuzumab result in reduction of fibrocyte differentiation. Treatment of a romiplostim-induced myelofibrotic mouse model with elotuzumab resulted in decreased BM fibrosis and reduction in spleen size. This serves as preclinical evidence that elotuzumab may modulate the BM microenvironment in MF patients and provide a more hospitable environment for normal hematopoiesis. (C) Elotuzumab targets the BM microenvironment through inhibiting differentiation of SLAMF7highCD16– to neoplastic fibrocytes; however, it is unlikely that this agent has any effect on the MPN hematopoietic stem cells. A combination therapy approach targeting the MPN hematopoietic stem cells and restoring the BM microenvironment would likely be the most effective strategy to achieve disease course modification.
The 2 main cellular drivers of BM fibrosis in MF are mesenchymal stromal cells and fibrocytes. Mesenchymal stromal cells are not derived from the neoplastic clone and are considered reactive mediators of collagen deposition.2,3 Fibrocytes arise from neoplastic monocytes, are mediators of reticulin deposition, and have been shown to stimulate the growth of normal and neoplastic hematopoietic colonies.4 Maekawa et al show an increase in the percentage of peripheral blood SLAMF7highCD16– monocytes in JAK2V617F+ MF patients compared with patients who have myeloproliferative neoplasms (MPNs) but do not have MF and healthy controls. This monocyte percentage was found to positively correlate with JAK2V617F allele burden. Using an in vitro culture assay, the authors show that treatment with the anti-SLAMF7 antibody elotuzumab can inhibit differentiation of neoplastic monocytes to fibrogenic fibrocytes. In addition, elotuzumab treatment of a romiplostim-induced myelofibrotic mouse model resulted in reduction in BM fibrosis and splenomegaly (see figure panels A-B).
MF is a clonal hematopoietic stem cell malignancy that is heterogeneous in clinical presentation and disease course but retains several hallmark biological features, including BM fibrosis, megakaryocyte hyperplasia, and constitutive activation of the JAK/STAT pathway. The exact mechanisms underlying disease pathogenesis and progression are unclear but likely involve the acquisition of somatic mutations, a proinflammatory milieu, and a dysfunctional BM microenvironment. Allogeneic hematopoietic stem cell transplantation remains the only potential curative option for this chronic and progressive myeloid neoplasm, which is feasible for only a minority of patients. Ruxolitinib (Jakafi, Incyte), a JAK1/2 inhibitor, is approved for the treatment of patients with intermediate-risk or high-risk MF and affords patients significant improvement in disease-related symptoms and splenomegaly. However, hematologic, BM morphologic, and molecular responses are generally not achieved with ruxolitinib therapy because of limited anticlonal activity. Patients for whom ruxolitinib therapy fails have a poor outcome, so treatments that can offer modification of the disease course with extension of survival are urgently needed.5
The results from the study by Maekawa et al are noteworthy and serve as preclinical evidence that inhibition of SLAMF7 signaling may modulate the MF BM microenvironment. There are limitations, however, which should be considered in the study itself and in the broader aspect of targeting the BM microenvironment. The murine romiplostim-induced model of MF used in this study is not a conventional MPN model like the GATA1low or MPL-mutant MPN murine models, and this work should be extended to a more MPN-relevant exemplar.
Although, the degree of BM fibrosis in MF has been correlated with overall survival,6 the consequence of therapeutic reversal of BM fibrosis is unclear, and clinical investigation has demonstrated only limited success. It is unlikely that correction of BM microenvironmental abnormalities alone will cure MF, and strategies aimed at eliminating the disease-initiating MPN hematopoietic stem cell are needed. The preclinical studies reported here illustrate the potential effects of elotuzumab on the BM niche, but whether there is a meaningful impact on the MPN hematopoietic stem cell is unclear. Because of the absence of SLAMF7 expression on CD34+ hematopoietic stem cells, it is unlikely that direct anticlonal activity will be seen with elotuzumab.7
It seems to be the case in multiple myeloma, but elotuzumab monotherapy is not clinically active unless it is used in combination with antiplasma cell-directed agents.8 Combining antifibrotic therapy with MPN hematopoietic stem cell–targeted therapy would likely be the most effective treatment strategy in MF to disarm and delete the malignant stem cells. Combination of elotuzmab or other antifibrotic agents such as the transforming growth factor β (TGF-β) ligand trap, AVID200 (NCT03895112), which is already in clinical testing may be ideally combined with rational partners that have laboratory evidence of activity against the MPN stem cell, such as MDM2 antagonists9 or bromodomain inhibitors10 (see figure panel C). Ultimately, it remains to be seen whether an antifibrotic agent such as elotuzumab will be effective in treating MF; however, without simultaneously targeting the MPN stem cell, there is low likelihood it will be a slam dunk as monotherapy.
Conflict-of-interest disclosure: J.M. reports that Icahn School of Medicine at Mount Sinai received funding from Incyte, Roche, Novartis, CTI Biopharma, Janssen, Merck, Promedior, and Celgene for clinical trial research. He is a clinical trial steering committee and scientific advisory board member for Roche, CTI Biopharma, Incyte, and Celgene. B.K.M. declares no competing financial interests.
