

Key Points
LMO2 is deacetylated by the NAMPT/SIRT2 pathway.
LMO2 deacetylation is essential for LIM domain binding 1 binding and TAL1 complex activation during hematopoiesis and T-ALL leukemogenesis.

Abstract
Hematopoietic transcription factor LIM domain only 2 (LMO2), a member of the TAL1 transcriptional complex, plays an essential role during early hematopoiesis and is frequently activated in T-cell acute lymphoblastic leukemia (T-ALL) patients. Here, we demonstrate that LMO2 is activated by deacetylation on lysine 74 and 78 via the nicotinamide phosphoribosyltransferase (NAMPT)/sirtuin 2 (SIRT2) pathway. LMO2 deacetylation enables LMO2 to interact with LIM domain binding 1 and activate the TAL1 complex. NAMPT/SIRT2-mediated activation of LMO2 by deacetylation appears to be important for hematopoietic differentiation of induced pluripotent stem cells and blood formation in zebrafish embryos. In T-ALL, deacetylated LMO2 induces expression of TAL1 complex target genes HHEX and NKX3.1 as well as LMO2 autoregulation. Consistent with this, inhibition of NAMPT or SIRT2 suppressed the in vitro growth and in vivo engraftment of T-ALL cells via diminished LMO2 deacetylation. This new molecular mechanism may provide new therapeutic possibilities in T-ALL and may contribute to the development of new methods for in vitro generation of blood cells.
Introduction
Hematopoietic transcription factors that play crucial roles during different stages of blood development are often deregulated in leukemia,1,-3 strengthening the view that appropriate regulation of transcription factor networks is essential for maintaining proper hematopoietic tissue homeostasis. One example of the importance of dose and cell differentiation stage-dependent expression of transcription factors in blood homeostasis is the LIM domain only 2 (LMO2) protein, an essential transcriptional regulator of early hematopoiesis.4,5 LMO2 knockout mice and zebrafish exhibit a complete loss of hematopoietic cells.6,7 Notably, malignant cells from ∼50% of patients with T-cell acute lymphoblast leukemia (T-ALL) express elevated levels of LMO2 or its interaction partner SCL/T-cell acute lymphocytic leukemia 1 (TAL1).8,-10 LMO2 is continuously silenced after commitment to early T-cell progenitors, and its overexpression leads to preleukemic alterations in thymocytes that culminate in T-ALL.11,,,-15 It has been shown that, in T-ALL, LMO2 reactivates a hematopoietic stem cell (HSC)-specific transcriptional program, leading to enhanced self-renewal and proliferation of early T-cell progenitors with reduced capacity for T-cell differentiation of T-ALL blasts.16 A recent study by García-Ramírez et al demonstrated that the presence of LMO2 in murine hematopoietic stem/progenitor cells (HSPC) is necessary for the early stages of transformation to T-ALL through in vivo reprogramming.17
LMO2, which is highly conserved in organisms ranging from zebrafish to humans,5 consists of 2 LIM domains (LIM1 and LIM2) connected by a short, flexible hinge region.18,19 LIM domains are generally composed of 2 consecutive zinc finger motifs that mediate interactions with other proteins. The LMO2 protein forms the core of the transcriptional TAL1 complex, anchoring its interaction partners LIM domain binding 1 (LDB1), TAL1 (also known as SCL), E47 (also known as transcription factor-3), and GATA binding protein 1 (GATA1).20 Both LMO2 LIM domains serve as scaffolds for assembly of the complex. Whereas the interaction with LDB1 involves all 4 zinc fingers, the interaction with the TAL1:E47 heterodimer is largely localized to the central hinge region, involving the C-terminal zinc finger of LIM1 and the N-terminal zinc finger of LIM2.19 GATA proteins are thought to interact mostly with the LIM2 domain.18 Thus, LMO2 functions as an essential adapter protein, allowing the proper assembly of the TAL1 complex.
Determining how LMO2 activity may be specifically targeted in T-ALL requires an understanding of the mechanisms of LMO2 activation. There are only a few reports describing the mechanism of LMO2 activation. Two of them demonstrated autoregulatory mechanisms of elevated LMO2 messenger RNA (mRNA) expression in HSCs and T-ALL cells.21,22 Posttranslational regulation of protein function through deacetylation mediated by nicotinamide phosphoribosyltransferase (NAMPT) and sirtuin (SIRT) is known to play a pivotal role during myeloid differentiation and leukemogenic transformation of hematopoietic cells. The NAMPT/SIRT pathway serves this function by activating a number of proteins, including the CCAAT/enhancer binding proteins C/EBPα and C/EBPβ, the serine/threonine kinase AKT, the tumor-suppressor p53, and the forkhead box transcription factor FOXO3.23,,,-27 NAMPT is a NAD+-generating enzyme, and SIRT family proteins (SIRT1-7) are NAD+-dependent class III histone deacetylases.28 Despite their high similarity, SIRT1 and SIRT2 have different functions, targets, and preferential intracellular localizations. In this latter context, SIRT1 is preferentially localized to the nucleus, whereas SIRT2 is a cytoplasmic enzyme that transiently migrates to the nucleus during the G2/M cell-cycle transition.29,30 It has been demonstrated that a SIRT1 deficiency compromises hematopoietic differentiation of mouse embryonic stem (ES) cells, and embryonic and adult hematopoiesis in the mouse.31 However, the role of NAMPT and SIRT2 during early stages of blood cell development or T-ALL leukemogenesis is largely unknown. Importantly, specific selective inhibitors of NAMPT, SIRT1, and SIRT2 have been described,32,-34 allowing functional analysis of the specific role of these factors. In the present study, we investigated whether LMO2 is activated by NAMPT/SIRT2-mediated deacetylation in T-ALL and in early blood development in vitro and in vivo.
Methods
Cell culture
HEK 293T and HEK 293FT cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. MOLT-4 and MOLT-14 cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. Mononuclear cells isolated from bone marrow or peripheral blood samples of 3 pediatric T-ALL patients were transplanted IV into 8- to 12-week-old female NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory, Bar Harbor, ME). T-ALL blasts isolated from spleen of engrafted mice were used for the in vitro experiments and the in vivo zebrafish engraftment. Written informed consent and institutional review board approval of the University Hospital Tübingen in accordance with the Declaration of Helsinki were obtained for the use of T-ALL bone marrow and peripheral blood samples for this study.
Healthy donor-derived human iPS cells (hCD34-iPSC16)35 were kindly provided by Nico Lachmann and Thomas Moritz (Hannover Medical School, Hannover, Germany). This human iPS cell line was maintained on mitomycin-C-treated (Sigma-Aldrich, St. Louis, MO) SNL feeder cells in Dulbecco’s modified Eagle medium/F12 (Sigma-Aldrich) supplemented with 20% Knockout Serum Replacement (Thermo Fisher Scientific, Waltham, MA), 30 ng/mL basic fibroblast growth factor (Peprotec, Rocky Hill, NJ), 1% nonessential amino acids solution (Thermo Fisher Scientific), 100 mM 2-mercaptethanol (Thermo Fisher Scientific), and 2 mM l-glutamine (Thermo Fisher Scientific). The culture medium was replaced daily with fresh medium. Colonies were mechanically passaged onto new SNL feeder cells every 10 days.
Hematopoietic differentiation of iPS cells
Undifferentiated iPS cell colonies were cultured on growth factor-reduced Matrigel (Becton-Dickinson)-coated cell culture dishes in Stemline II HSC expansion medium (Sigma-Aldrich) containing insulin-transferrin-selenium (ITS) supplement (Thermo Fisher Scientific) and cytokines (bone morphogenetic protein 4 [BMP4], 20 ng/mL, R&D Systems, Minneapolis, MN) was added to the culture for the first 4 days and then replaced with vascular endothelial growth factor 165 (VEGF 165) (40 ng/mL, R&D Systems) for 2 days. On day 6, VEGF 165 was replaced with a combination of stem cell factor (50 ng/mL, R&D Systems), interleukin 3 (IL-3; 50 ng/mL, R&D Systems), and thrombopoietin (5 ng/mL, R&D Systems). Thereafter, medium was replaced every 3 or 4 days. FK866 (Sigma-Aldrich), AC93253 (Sigma-Aldrich), EX527 (Sigma-Aldrich), or dimethyl sulfoxide (DMSO; vehicle control, Sigma-Aldrich) were added to the fresh medium. Differentiated iPS cells were dissociated using StemPro Accutase Cell Dissociation Reagent (Thermo Fisher Scientific), harvested through a cell strainer (Miltenyi Biotec, Bergisch Gladbach, Germany), and used for analysis. CD34+ cells were sorted using the CD34 MicroBead Kit, human (Miltenyi Biotec) following the manufacturer’s instructions.
Zebrafish experiments
Gene knockdown experiments were performed using wild-type (WT) TE fish and the transgenic line Tg(-6.35drl:EGFP).36 Antisense morpholino oligonucleotides (MOs) were obtained from GENE TOOLS (Philomath, OR) targeting nampta (5′-TGTGTGACCTGCAATGAAAGAAAGA-3′) or sirt237 (5′-ACCTCTAAAGGACACAAAAAAGGCT-3′). MOs and plasmids (pcDNA3.1-pbef [NAMPT], pcDNA3.1-SIRT2-flag, and pSIN4-EF1a-LMO2-IRES-Puro, with WT or K74/78R mutant LMO2 complementary DNA) were injected into 1-cell stage embryos using a PV830 Pneumatic PicoPump (World Precision Instruments, Sarasota, FL) at nontoxic levels (ie, 3.3 ng nampta MO, 2.5 ng sirt2 MO and 25 pg plasmids). Primers flanking exon 1 and exon 5 of the nampta gene (forward: 5′-AGAGAAGCCGCGGATTTCAA-3′; reverse: 5′-CTCCAGTCCTTCCAGGCTTC-3′) and primers flanking exon 1 and exon 4 of the sirt2 gene37 (forward: 5′-GCTGGCTTATAGTTTTAAAGAGGGTA-3′; reverse: 5′-AGTATGTAGCGAGCAACTGAGTC-3′) were used for subsequent reverse transcriptase polymerase chain reaction (RT-PCR) to verify morpholino-induced blocking of splicing. Embryos at 24 hours postfertilization (hpf) were collected and homogenized with a pestle for RNA isolation and NAD+ measurement. RNA was isolated using the RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. Images were collected using an Axio ZoomV16 microscope (ZEISS).
Xenotransplantation experiments were performed using WT TE or albino fish (albb4).38 WT embryos were treated with 0.003% 1-phenyl-2-thiourea to prevent pigment formation. MOLT-4 cells were labeled with Vybrant CFDA SE Cell Tracer Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. Labeled cells were suspended in phosphate-buffered saline at a density of 2 × 105 cells/µL, and 1 nL cell suspension (200 cells) was injected into the perivitelline space of embryos at 24 hpf. Injected embryos were incubated at 28°C for 1 hour followed by incubation with DMSO or FK866 for 48 hours at 35°C. Drug-treated embryos were dissociated by passing through a 40-µm cell strainer (Greiner Bio-One) in ice-cold FACS buffer (phosphate-buffered saline, 2% FCS, 0.05% NaN3) and then analyzed using a BD LSR II Flow Cytometer (Becton-Dickinson) and FlowJo software (FlowJo, LLC).
Xenotransplantation in mice
NSG mice (The Jackson Laboratory) were maintained under specific pathogen-free conditions in the research animal facility of the University of Tübingen, Germany, according to German federal and state regulations (Regierungspräsidium Tübingen, K3/17). Primary T-ALL patient cells or MOLT-4 cells (0.5 × 106 cells each) were injected IV into 8- to 12-week old male NSG mice and randomized to the treatment groups. Seven days after leukemia inoculation, mice were injected intraperitoneally once a day with 20 mg/kg FK866 hydrochloride hydrate (dissolved in 20% [2-Hydroxypropyl]-γ-cyclodextrin [HGC]) or 20% HGC, respectively. Two or 3 weeks after starting the treatment, mice were euthanized, and the bone marrow was analyzed for human CD45+ leukemia cells by flow cytometry. Engrafted T-ALL cells were isolated from bone marrow samples using the human CD45 MicroBeads Kit (Miltenyi Biotec) following the manufacturer’s instructions.
Statistical analyses
Statistical analyses were conducted using Student t test or BootstRatio.39 Statistical significance was taken to be P < .05.
Results
Lysine deacetylation of LMO2 is essential for the binding to LDB1 protein and activation of the TAL1 complex
Using the in silico prediction algorithms PAIL,40 LysAcet41 and LAceP,42 we identified 2 evolutionarily highly conserved lysine residues, K74 and K78, in the LIM1 domain of LMO2 that may be targets for deacetylation (Figure 1A). Acetylation of LMO2 protein was confirmed in immunoprecipitation assay using HEK293T cells, in which acetylation of LMO2 protein was enhanced by cotransfection with the lysine acetyltransferase p300 (Figure 1B). We confirmed acetylation of the endogenous LMO2 protein in the LMO2-expressing T-ALL cell line MOLT4 in immunoprecipitation assay (supplemental Figure 1A-B, available on the Blood Web site).
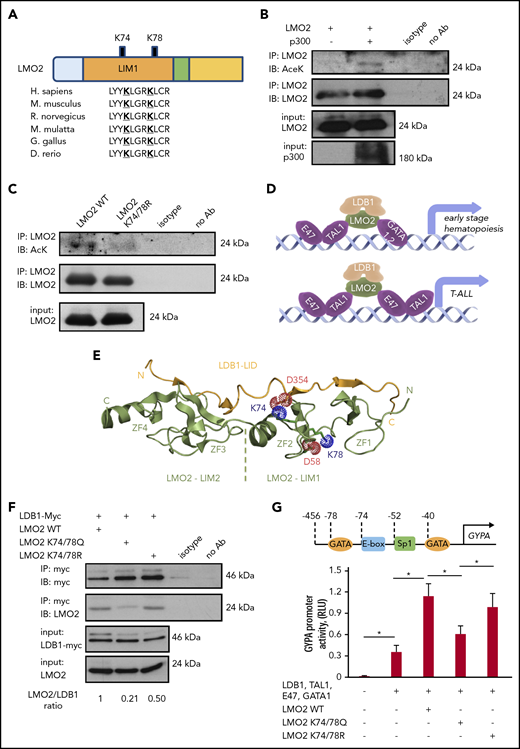
Lysine deacetylation of LMO2 is essential for the LDB1 interaction and transcriptional activity of the TAL1 complex. (A) Schematic of the LMO2 protein showing 2 putative lysines in LMO2 (K74 and K78) within the LIM1 domain that could be deacetylated (bold and underlined) based on in silico analysis. (B-C) Immunoprecipitation (IP) assay of the lysates of HEK293T cells transfected with expression vectors of p300 and WT or K74/78R LMO2 mutant. IP and immunoblot were performed with indicated antibodies. IPs were performed twice; representative western blot images are depicted. (D) Schematic of the activation of target genes during early hematopoiesis (upper) and T-ALL leukemogenesis (lower) by binding of the TAL1 complex (consisting of E47, TAL1, LDB1, LMO2, and GATA1/2 proteins) to the specific putative regions on the gene promoters. (E) Crystal structure of the LMO2:LDB1-LID complex (PDB 2XJY), highlighting electrostatic interactions of the deacetylated lysines 74 and 78 of LMO2 (green). The lysines and neighboring aspartates, D58 from LMO2-LIM1 and D354 from LDB1 (yellow), are shown as sticks; their cationic ammonium and anionic carboxylate groups are outlined with dotted spheres representing their van der Waals radii. If the lysines were acetylated, they would carry 0 net-charge and could not complement the negative charge of the aspartates. N and C termini and the individual zinc fingers (ZF1-ZF4) are labeled. (F) Co-IP of LMO2 and LDB1 proteins in lysates of HEK293T cells transfected with indicated expression vectors. IP and immunoblot were performed with indicated antibodies. Representative western blot images are depicted. (G) Activity of GYPA reporter containing the 456-bp upstream region of GYPA with putative binding sites for the TAL1 complex (upper). HEK293T cells were transfected with GYPA reporter construct and the indicated expression vectors. The activation of GYPA reporter was measured as described in “Methods." Data show means ± standard deviation (SD) (n = 8). *P < .05.
Lysine deacetylation of LMO2 is essential for the LDB1 interaction and transcriptional activity of the TAL1 complex. (A) Schematic of the LMO2 protein showing 2 putative lysines in LMO2 (K74 and K78) within the LIM1 domain that could be deacetylated (bold and underlined) based on in silico analysis. (B-C) Immunoprecipitation (IP) assay of the lysates of HEK293T cells transfected with expression vectors of p300 and WT or K74/78R LMO2 mutant. IP and immunoblot were performed with indicated antibodies. IPs were performed twice; representative western blot images are depicted. (D) Schematic of the activation of target genes during early hematopoiesis (upper) and T-ALL leukemogenesis (lower) by binding of the TAL1 complex (consisting of E47, TAL1, LDB1, LMO2, and GATA1/2 proteins) to the specific putative regions on the gene promoters. (E) Crystal structure of the LMO2:LDB1-LID complex (PDB 2XJY), highlighting electrostatic interactions of the deacetylated lysines 74 and 78 of LMO2 (green). The lysines and neighboring aspartates, D58 from LMO2-LIM1 and D354 from LDB1 (yellow), are shown as sticks; their cationic ammonium and anionic carboxylate groups are outlined with dotted spheres representing their van der Waals radii. If the lysines were acetylated, they would carry 0 net-charge and could not complement the negative charge of the aspartates. N and C termini and the individual zinc fingers (ZF1-ZF4) are labeled. (F) Co-IP of LMO2 and LDB1 proteins in lysates of HEK293T cells transfected with indicated expression vectors. IP and immunoblot were performed with indicated antibodies. Representative western blot images are depicted. (G) Activity of GYPA reporter containing the 456-bp upstream region of GYPA with putative binding sites for the TAL1 complex (upper). HEK293T cells were transfected with GYPA reporter construct and the indicated expression vectors. The activation of GYPA reporter was measured as described in “Methods." Data show means ± standard deviation (SD) (n = 8). *P < .05.
To evaluate the role of K74 and K78 of LMO2 on its deacetylation, we generated expression constructs containing WT or deacetylation-mimic LMO2 mutant at the predicted lysine residues (K74/78R). Immunoprecipitation assays in HEK293T cells cotransfected with LMO2 constructs and p300 using anti-LMO2 antibody followed by western blot with anti-acetyl-K antibody confirmed acetylation of WT LMO2 and demonstrated that deacetylation-mimic LMO2 mutant at K74/78R could not be acetylated (Figure 1C). LMO2 is a component of the multiprotein transcriptional activation TAL1 complex, consisting of LMO2, LDB1, TAL1, E47, and GATA1/2 proteins that exert transcriptional activity toward target genes activated during early hematopoiesis and in T-ALL blasts (Figure 1D).43 Therefore, we aimed to investigate whether LMO2 deacetylation on K74 and K78 may affect its interaction with LDB1. Structurally, both K74 and K78 are accommodated in the second zinc finger of LMO2, but only K74 is directly involved in the interaction with LDB1. The association of LMO2 and LDB1 is driven by both polar and hydrophobic contacts. The LDB1-LID domain binds in an elongated conformation along the 2 LMO2 LIM domains by contributing an additional β-strand to each of the 4 zinc finger domains (Figure 1E). Consequently, a large network of backbone hydrogen bonds is formed in these extended β-sheets, which are fortified by several hydrophobic contacts between the LID and both LIM domains. However, there are also several electrostatic interactions, which are exclusively formed via the LIM1 domain.18 In addition to the described salt bridges between R40, E52, and R70 of LIM1 and E362, R360, and E355 of LDB1-LID, we note that K74 is also in direct van der Waals distance to D354 for an ionic interaction. Together with D352, E353, and E355, D354 forms a strongly anionic DEDE motif, which is counterbalanced by the cationic R70 and K74. Consequently, when K74 is acetylated (and thus neutralized), the interaction with D354 is lost and the electrostatic balance between LMO2 and LDB1 impaired. In contrast, K78 is not directly interacting with LDB1-LID but forms a salt bridge with D58 of the same zinc finger (Figure 1E). It is conceivable that this interaction is stabilizing the geometry of the LIM domain. This notion is supported by the observation that in 1 instance of a LMO2:LDB1-LID crystal structure (PDB 2XJZ, chain C), in which this interaction is broken, the geometry of LIM1 is distorted and the LDB1-LID domain detached from the first zinc finger.18 It is therefore conceivable that the acetylation of K78 would lead to a similar conformational change and thus abrogate the LMO2:LDB1 interaction.
We compared the interaction between LDB1 and WT K74/78Q (acetylation-mimic) or K74/78R (deacetylation-mimic) LMO2 expression constructs by co-immunoprecipitation and indeed found that the K74/78Q LMO2 mutant showed a weaker interaction with LDB1 in comparison with WT and K74/78R LMO2 (Figure 1F).
Furthermore, we made a reporter gene construct containing a 456-bp upstream regulatory region of the human GYPA gene containing GATA binding motifs, an E-box and SP1 binding site specific for TAL1 complex binding44 and performed reporter gene assay. We found that the acetylation-mimic K74/78Q LMO2 mutant suppressed GYPA promotor activity of the TAL1 complex in comparison with WT and the deacetylation-mimic K74/78R LMO2 mutant (Figure 1G). These results demonstrate that deacetylation of LMO2 at lysines 74 and 78 is essential for the transcriptional activity of the TAL1 complex.
Activation of LMO2 by NAMPT-mediated deacetylation
Proteins may be deacetylated by the NAMPT/SIRT pathway.45 NAMPT is essential for the generation of NAD+ and activation of NAD+-dependent protein deacetylases, SIRT (Figure 2A). To evaluate whether transcriptional activity of the TAL1 complex is affected by NAMPT-triggered LMO2 deacetylation, we compared TAL1 complex-dependent activation of the GYPA promoter in HEK293T cells in the presence or absence of the specific NAMPT inhibitor FK866. Indeed, we observed markedly diminished activation of TAL1 complex-dependent GYPA promoter activity after NAMPT inhibition compared with DMSO-treated control group (Figure 2B).
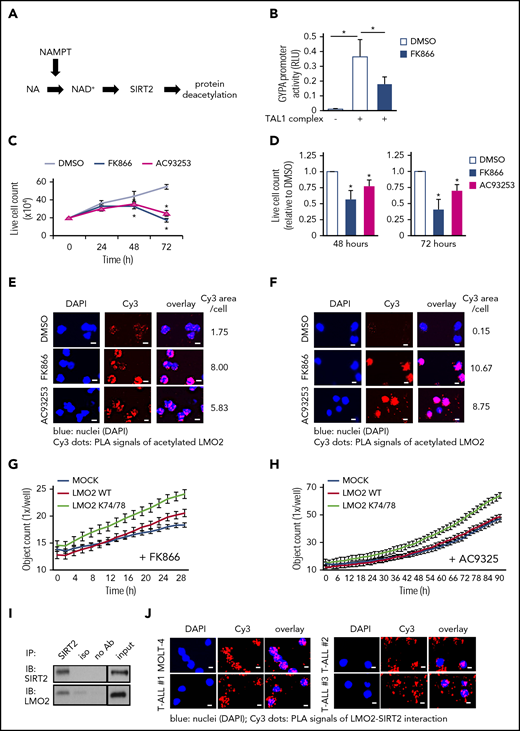
The NAMPT/SIRT2 pathway activates LMO2 by deacetylation in T-ALL cells. (A) Schematic of the NAMPT-NAD+-SIRTs pathway. (B) Reporter gene assay using GYPA reporter construct. HEK293T cells were transfected with a GYPA reporter and the indicated expression constructs. Transfected cells were cultured in the presence or absence of FK866 for 24 hours, and the activation of the GYPA reporter was measured as described in “Methods." The same concentration of DMSO was added as a vehicle control. Data show means ± SD (n = 8). *P < .05. (C) Live-cell counting of MOLT-4 cells treated with 10 nM FK866 or 100 nM AC93253. The same concentration of DMSO was added as a vehicle control. Dead cells were excluded by trypan blue dye staining. Data represent mean ± SD from 3 independent experiments, each in triplicate (*P < .05 compared with DMSO-treated cells). (D) Live-cell counting of primary blasts from T-ALL patients (n = 3) treated with 10 nM FK866 or 100 nM AC93253 for 48 or 72 hours. The same concentration of DMSO was added as a vehicle control. Dead cells were excluded by trypan blue dye staining. Live-cell numbers are normalized to those obtained in samples treated with DMSO (defined as 1). Data represent means ± SD of triplicate determinations (*P < .05 compared with DMSO-treated cells). Duolink in situ proximity ligation assay (PLA) of MOLT-4 cells (E) or primary blasts (F) from T-ALL patient (patient no. 3) treated with 10 nM FK866 or 100 nM AC93253 for 48 hours using anti-LMO2 and anti-acetylated lysine antibodies. The same concentration of DMSO was added as a vehicle control. Representative images are shown. Scale bars: 10 μm. (G-H) Proliferation of MOCK, WT-LMO2, or K74/78R LMO2-expressing MOLT4 cells (2 × 104 cells/well of a 96-well plate) was evaluated over time with the IncuCyte S3 Live-Cell Analysis System. The experiment was performed twice, each in triplicate. (I) Co-IP assay of SIRT2 and LMO2 protein interaction in lysates of HEK293T cells transfected with SIRT2 and LMO2 expression vectors. IP and immunoblotting were performed using the indicated antibodies. Representative images are shown. (J) Duolink in situ PLAs in MOLT-4 cells and primary blasts of T-ALL patients. Cells were stained with anti-LMO2 and anti-SIRT2 antibodies. Representative images are shown. Scale bars: 10 μm. RLU, relative light unit.
The NAMPT/SIRT2 pathway activates LMO2 by deacetylation in T-ALL cells. (A) Schematic of the NAMPT-NAD+-SIRTs pathway. (B) Reporter gene assay using GYPA reporter construct. HEK293T cells were transfected with a GYPA reporter and the indicated expression constructs. Transfected cells were cultured in the presence or absence of FK866 for 24 hours, and the activation of the GYPA reporter was measured as described in “Methods." The same concentration of DMSO was added as a vehicle control. Data show means ± SD (n = 8). *P < .05. (C) Live-cell counting of MOLT-4 cells treated with 10 nM FK866 or 100 nM AC93253. The same concentration of DMSO was added as a vehicle control. Dead cells were excluded by trypan blue dye staining. Data represent mean ± SD from 3 independent experiments, each in triplicate (*P < .05 compared with DMSO-treated cells). (D) Live-cell counting of primary blasts from T-ALL patients (n = 3) treated with 10 nM FK866 or 100 nM AC93253 for 48 or 72 hours. The same concentration of DMSO was added as a vehicle control. Dead cells were excluded by trypan blue dye staining. Live-cell numbers are normalized to those obtained in samples treated with DMSO (defined as 1). Data represent means ± SD of triplicate determinations (*P < .05 compared with DMSO-treated cells). Duolink in situ proximity ligation assay (PLA) of MOLT-4 cells (E) or primary blasts (F) from T-ALL patient (patient no. 3) treated with 10 nM FK866 or 100 nM AC93253 for 48 hours using anti-LMO2 and anti-acetylated lysine antibodies. The same concentration of DMSO was added as a vehicle control. Representative images are shown. Scale bars: 10 μm. (G-H) Proliferation of MOCK, WT-LMO2, or K74/78R LMO2-expressing MOLT4 cells (2 × 104 cells/well of a 96-well plate) was evaluated over time with the IncuCyte S3 Live-Cell Analysis System. The experiment was performed twice, each in triplicate. (I) Co-IP assay of SIRT2 and LMO2 protein interaction in lysates of HEK293T cells transfected with SIRT2 and LMO2 expression vectors. IP and immunoblotting were performed using the indicated antibodies. Representative images are shown. (J) Duolink in situ PLAs in MOLT-4 cells and primary blasts of T-ALL patients. Cells were stained with anti-LMO2 and anti-SIRT2 antibodies. Representative images are shown. Scale bars: 10 μm. RLU, relative light unit.
In vitro inhibition of NAMPT or SIRT2 suppresses proliferation and induces apoptosis of T-ALL cells by inhibiting LMO2 deacetylation
LMO2 and/or other components of the TAL1 complex are hyperactivated in T-ALL.46,47 Accordingly, we evaluated whether inhibition of NAMPT or SIRTs (SIRT1 and SIRT2) affects the proliferation and/or survival of the LMO2-expressing T-ALL cell lines, MOLT4 and MOLT14 (supplemental Figure 1A). We found that inhibition of NAMPT (FK866) or SIRT2 (AC93253), but not SIRT1 (EX527), led to a marked reduction in cell proliferation, as assessed by cell counts, 3H-thymidine incorporation, and BrdU assays, compared with vehicle (DMSO)-treated samples (Figure 2C; supplemental Figure 1C-H; data not shown). Apoptosis was also elevated in cells treated with FK866 or AC93253 (supplemental Figure 2A) compared with vehicle controls. These phenotypic changes are consistent with the almost completely abolished levels of intracellular NAD+ (supplemental Figure 2B). Strong dependence of MOLT4 cells on LMO2 was confirmed in CRISPR/Cas9-mediated LMO2 knockout (KO) studies, where we found dramatically diminished viability, proliferation, and elevated apoptosis of MOLT4 cells upon LMO2 KO (supplemental Figure 2C-H; supplemental Table 1).
We further evaluated the effects of NAMPT- or SIRT2 inhibition on the survival of primary blasts from T-ALL patients. We selected T-ALL patients expressing LMO2 and TAL1 levels comparable to that of the LMO2-dependent MOLT4 cell line having high LMO2 levels (supplemental Figure 1A; supplemental Table 2; supplemental Figure 3A). Treatment of LMO2- and TAL1-expressing primary blasts from T-ALL patients with FK866 or AC93253 also led to markedly diminished cell proliferation and reduced NAD+ levels (in FK866 treated cells) (Figure 2D; supplemental Table 2; supplemental Figure 3A-B). These findings are in line with the diminished deacetylation of LMO2 following treatment of the MOLT4 T-ALL cell line and primary T-ALL blasts with FK866 or AC93253 compared with cells in vehicle control groups (Figure 2E-F; supplemental Figure 3C). The specificity of the Duolink signal of acetylated LMO2 protein was confirmed in LMO2 KO MOLT4 cells (supplemental Figure 3D). Interestingly, FK866- or AC93253-treated MOLT4 cells stably expressing the K74/78R LMO2 mutant showed better proliferation compared with control transduced cells, as evaluated using an IncuCyte cell proliferation assay (Figure 2G-H).
We further evaluated whether SIRT2 interacts with LMO2 to induce its deacetylation. Indeed, direct interactions between SIRT2 and LMO2 were detected by coimmunoprecipitation and subsequent western blotting of lysates from HEK293T cells cotransfected with SIRT2 and LMO2 (Figure 2I), as well as in Duolink assays of T-ALL cell lines and primary cells from T-ALL patients (Figure 2J; supplemental Figure 4A). Moreover, interactions between endogenous LMO2 and LDB1 in MOLT-4 cells were considerably diminished by inhibition of NAMPT or SIRT2 (Figure 3A-B; supplemental Figure 4B), and TAL1 complex activity toward the GYPA reporter was reduced in SIRT2-KO HEK293FT cells (Figure 3C; supplemental Figure 3C-D). In addition, mRNA levels of the known LMO2-target genes, HHEX16 and NKX3.1,48 were strongly decreased in cells treated with FK866 or AC93253 (Figure 3D; supplemental Figure 4E). Consistent with the previously reported autoregulation of LMO2 expression in HSCs and T-ALL cells,21,22 LMO2 mRNA levels were reduced after NAMPT or SIRT2 inhibition (Figure 3D; supplemental Figure 4E).
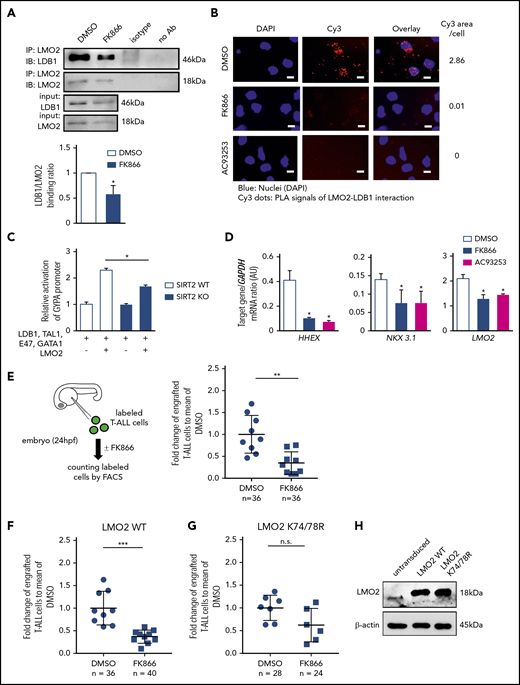
NAMPT/SIRT2-mediated LMO2 deacetylation is essential for LMO2 interaction with LDB1, activation of TAL-1 complex, and proliferation of T-ALL cells. (A) Co-IP of LMO2 and LDB1 from lysates of MOLT-4 cells treated with 10 nM FK866 for 72 hours. IP and immunoblotting were performed using the indicated antibodies. Representative images are shown. Bar graphs of the LDB1/LMO2 binding ratio indicate the LDB1 protein signal divided by the LMO2 protein signal in IP samples. Data show means ± SD from 3 independent experiments (*P < .05). (B) Duolink in situ PLAs of MOLT-4 cells treated with 10 nM FK866 or 100 nM AC93253 for 48 hours. Cells were stained with anti-LMO2 and anti-LDB1 antibodies. Scale bars: 10 μm. (C) WT or SIRT2-knockout (SIRT2KO) HEK293FT cells were transfected with a GYPA reporter and the indicated expression constructs. Transfected cells were cultured for 24 hours; activation of the GYPA reporter was measured as described in "Methods." Signals measured in cells cotransfected with LMO2 and GYPA constructs were normalized to those in cells without an LMO2 expression plasmid (defined as 1). Data show means ± SD (n = 8; *P < .05). (D) mRNA expression levels of the indicated genes in MOLT-4 cells treated with 10 nM FK866, 100 nM AC93253, or DMSO for 72 hours were assessed using quantitative RT-PCR. Target gene/GAPDH mRNA expression ratios are shown. Data, expressed as arbitrary units (AUs), are means ± SD from 3 independent experiments, each in triplicate (*P < .05 compared with DMSO-treated cells). (E-G) Xenotransplantation of T-ALL cells into zebrafish embryos. Zebrafish embryos were transplanted with labeled primary T-ALL cells (E), MOLT-4 cells transduced with a WT (F), or K74/78R mutant LMO2-expressing lentivirus construct (G). FACS, fluorescence-activated cell sorting. Xenotransplanted embryos were treated with 800 nM FK866 or DMSO for 2 days. Numbers of transplanted cells were assessed as described in "Methods." Data represent percentage of labeled cells normalized to the average percentage of labeled cells in the DMSO-treated group. The black lines represent mean ± SD. Each dot represents 4 embryos pooled as 1 biological sample. The number of analyzed embryos is indicated in the lower panel of each figure (***P < .0001; **P < .001). (H) Representative western blot images of cell lysates of MOLT-4 cells transduced with the indicated lentivirus constructs using anti-LMO2 and anti-β-actin antibodies. NS, not significant.
NAMPT/SIRT2-mediated LMO2 deacetylation is essential for LMO2 interaction with LDB1, activation of TAL-1 complex, and proliferation of T-ALL cells. (A) Co-IP of LMO2 and LDB1 from lysates of MOLT-4 cells treated with 10 nM FK866 for 72 hours. IP and immunoblotting were performed using the indicated antibodies. Representative images are shown. Bar graphs of the LDB1/LMO2 binding ratio indicate the LDB1 protein signal divided by the LMO2 protein signal in IP samples. Data show means ± SD from 3 independent experiments (*P < .05). (B) Duolink in situ PLAs of MOLT-4 cells treated with 10 nM FK866 or 100 nM AC93253 for 48 hours. Cells were stained with anti-LMO2 and anti-LDB1 antibodies. Scale bars: 10 μm. (C) WT or SIRT2-knockout (SIRT2KO) HEK293FT cells were transfected with a GYPA reporter and the indicated expression constructs. Transfected cells were cultured for 24 hours; activation of the GYPA reporter was measured as described in "Methods." Signals measured in cells cotransfected with LMO2 and GYPA constructs were normalized to those in cells without an LMO2 expression plasmid (defined as 1). Data show means ± SD (n = 8; *P < .05). (D) mRNA expression levels of the indicated genes in MOLT-4 cells treated with 10 nM FK866, 100 nM AC93253, or DMSO for 72 hours were assessed using quantitative RT-PCR. Target gene/GAPDH mRNA expression ratios are shown. Data, expressed as arbitrary units (AUs), are means ± SD from 3 independent experiments, each in triplicate (*P < .05 compared with DMSO-treated cells). (E-G) Xenotransplantation of T-ALL cells into zebrafish embryos. Zebrafish embryos were transplanted with labeled primary T-ALL cells (E), MOLT-4 cells transduced with a WT (F), or K74/78R mutant LMO2-expressing lentivirus construct (G). FACS, fluorescence-activated cell sorting. Xenotransplanted embryos were treated with 800 nM FK866 or DMSO for 2 days. Numbers of transplanted cells were assessed as described in "Methods." Data represent percentage of labeled cells normalized to the average percentage of labeled cells in the DMSO-treated group. The black lines represent mean ± SD. Each dot represents 4 embryos pooled as 1 biological sample. The number of analyzed embryos is indicated in the lower panel of each figure (***P < .0001; **P < .001). (H) Representative western blot images of cell lysates of MOLT-4 cells transduced with the indicated lentivirus constructs using anti-LMO2 and anti-β-actin antibodies. NS, not significant.
Inhibition of NAMPT results in diminished proliferation of T-ALL cells in vivo due to abrogated deacetylation of LMO2
We further evaluated the effect of NAMPT inhibition on the proliferation and survival of LMO2-expressing T-ALL cells in vivo. To this end, we injected MOLT-4 T-ALL cells or primary T-ALL patient cells labeled with the dye carboxyfluorescein diacetate succinimidyl ester into zebrafish embryos at 24 hpf and subsequently treated embryos with FK866 or DMSO for 48 hours. In line with our in vitro observations, the number of primary T-ALL patient cells in FK866-treated embryos was markedly reduced compared to control DMSO-treated samples (Figure 3E). In addition, transduction of MOLT-4 cells with a deacetylation-mimicking LMO2 (K74/K78), but not with WT LMO2, protected these cells from FK866-mediated cell death in vivo (Figure 3F-H).
In another series of experiments, we engrafted primary T-ALL patient cells and MOLT-4 cells into immunodeficient nonobese diabetic severe combined immunodeficiency IL2Rγc−/− (NSG) mice. Engrafted mice were treated with 20 mg/kg FK866 or HGC (solvent for FK866) for 14 (MOLT-4 cells) or 21 (primary T-ALL patient cells) days, once a day (Figure 4A-D). Similar to zebrafish xenograft experiments, the percentage of engrafted T-ALL cells was strongly reduced in FK866-treated mice compared with HGC-injected control animals (Figure 4 B-C). LMO2 deacetylation was reduced and interactions between LMO2 and LDB1 were markedly diminished in T-ALL cells isolated from the bone marrow of engrafted FK866-treated mice (Figure 4E-H). mRNA expression levels of HHEX and NKX3.1 were reduced in MOLT4 cells engrafted in FK866-treated mice compared with that observed in HCG-treated mice (Figure 4I). Again, transduction of cells with the deacetylation-mimicking K74/78R LMO2 mutant, but not WT LMO2 (as control) prevented FK866-mediated abrogation of engraftment (Figure 4C-D), and rescued downregulation of LMO2 target genes (Figure 4I). In line with this, LMO2 deacetylation was reduced and interactions between LMO2 and LDB1 were much weaker in LMO2-WT-transduced MOLT4 cells isolated from the bone marrow of engrafted FK866-treated mice, but not in cells transduced with the K74/K78 LMO2 mutant (Figure 4G-H).
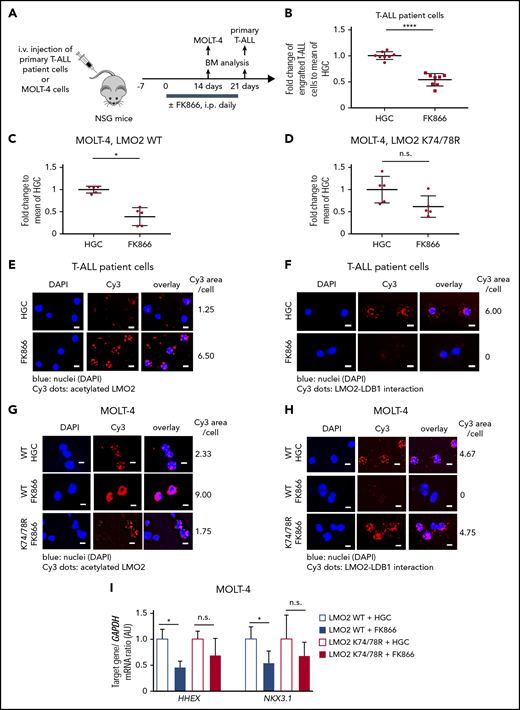
In vivoinhibition of NAMPT results in reduced T-ALL burden in NSG mice owing to abrogated deacetylation of LMO2. (A) Xenotransplantation of NSG mice with either MOLT-4 cells, transduced with WT or K74/78R mutant LMO2-lentivirus, or primary T-ALL patient cells. Transplanted mice were injected intraperitoneally (i.p.) with 20 mg/kg of FK866 once a day for 2 (MOLT4 cells) or 3 (T-ALL patient cells) weeks. The same concentration of HGC was injected as a vehicle control. (B-D) Fold changes of percent of human CD45+ primary T-ALL patient cells (B) and Molt-4 cells (C-D) in bone marrow of FK866-treated mice to HGC group are shown. Data represent mean ± standard error of the mean (****P < .0001), n = 5 to 8 mice per group. (E-H) Duolink in situ PLAs of human CD45+ cells isolated from bone marrow of mice transplanted with primary T-ALL patient cells (E-F) or MOLT-4 cells (G-H). “WT” and “K74/78R” indicate mice transplanted with MOLT-4 cells transduced with WT or K74/48R LMO2 mutant, respectively. Cells were stained with anti-LMO2 and anti-acetylated lysine (E,G) or anti-LMO2 and anti-LDB1 antibodies (F,H). Representative images are shown. Scale bars: 10 μm. (I) mRNA expression levels of the indicated genes in human CD45+ cells isolated from bone marrow of xenotransplanted mice. mRNA expression was assessed using quantitative RT-PCR, and is expressed as arbitrary units (AUs). Target gene/GAPDH mRNA expression ratios are shown. Data show means ± SD from 5 independent experiments, each in triplicate (*P < .05).
In vivoinhibition of NAMPT results in reduced T-ALL burden in NSG mice owing to abrogated deacetylation of LMO2. (A) Xenotransplantation of NSG mice with either MOLT-4 cells, transduced with WT or K74/78R mutant LMO2-lentivirus, or primary T-ALL patient cells. Transplanted mice were injected intraperitoneally (i.p.) with 20 mg/kg of FK866 once a day for 2 (MOLT4 cells) or 3 (T-ALL patient cells) weeks. The same concentration of HGC was injected as a vehicle control. (B-D) Fold changes of percent of human CD45+ primary T-ALL patient cells (B) and Molt-4 cells (C-D) in bone marrow of FK866-treated mice to HGC group are shown. Data represent mean ± standard error of the mean (****P < .0001), n = 5 to 8 mice per group. (E-H) Duolink in situ PLAs of human CD45+ cells isolated from bone marrow of mice transplanted with primary T-ALL patient cells (E-F) or MOLT-4 cells (G-H). “WT” and “K74/78R” indicate mice transplanted with MOLT-4 cells transduced with WT or K74/48R LMO2 mutant, respectively. Cells were stained with anti-LMO2 and anti-acetylated lysine (E,G) or anti-LMO2 and anti-LDB1 antibodies (F,H). Representative images are shown. Scale bars: 10 μm. (I) mRNA expression levels of the indicated genes in human CD45+ cells isolated from bone marrow of xenotransplanted mice. mRNA expression was assessed using quantitative RT-PCR, and is expressed as arbitrary units (AUs). Target gene/GAPDH mRNA expression ratios are shown. Data show means ± SD from 5 independent experiments, each in triplicate (*P < .05).
Gene expression signature of NAMPT-deacetylated LMO2 in T-ALL
To identify the gene expression program regulated by NAMPT-deacetylated LMO2 in T-ALL, we performed RNA sequencing (RNA-Seq) analysis of WT or K74/78 LMO2-expressing MOLT4 cells treated with FK866 or DMSO for 2 days (Figure 5A). We detected 69 genes to be differentially regulated in WT LMO2-, but not K74/78 LMO2-expressing cells after NAMPT inhibition by FK866 (Figure 5B-C; supplemental Table 3). Among these genes, we found tumor- or leukemia-associated candidates, as PRDM1, HOXD13, RASL11B, TRAF1, IGFBP4, SOX7, SOX12, CYP1B1, and CYP2E1 (Figure 5C). Pathway overrepresentation analysis49 of the candidate genes revealed cytochrome P450, tryptophan degradation, biological oxidation, direct p53 effectors, and tumor necrosis factor α (TNF-α) signaling pathways to be regulated by NAMPT-deacetylated LMO2 (Figure 5D). Using iRegulon analysis of the transcription factor binding sites50 presented in FK866-treated WT LMO2, but not in K74/78 LMO2 MOLT4 cells, as compared with each DMSO control, we detected PITX-, ZNF695-, MEF2-, and Ep300-binding motifs to be significantly enriched (Figure 5E).
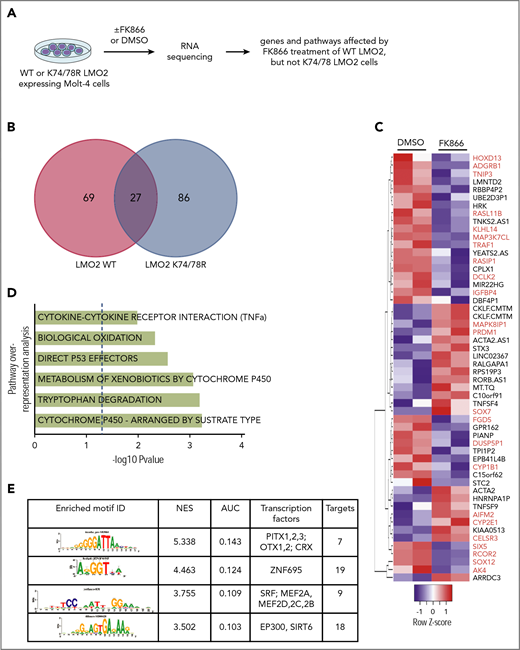
NAMPT-LMO2 dependent gene expression signature in MOLT4 cells. (A) Schematic of the experimental procedure. Venn diagram (B) and heat map (C) of genes that were significantly differentially regulated upon FK866 treatment compared with DMSO-treated groups of WT LMO2 or K74/78 LMO2 expressing MOLT4 cells (adjusted value P < .05). Genes known to play a role in tumorigenesis are marked in red. (D) Pathway overrepresentation analysis using a list of significantly regulated genes from panel C revealed top significantly enriched signal transduction pathways that are NAMPT-LMO2 dependent. (E) iRegulon transcription factor motif enrichment analysis using a list of significantly upregulated genes from (C) revealed enrichment of genes (normalized enrichment score >3.0 and area under the curve >0.03) with NAMPT-LMO2–specific transcription factor motifs.
NAMPT-LMO2 dependent gene expression signature in MOLT4 cells. (A) Schematic of the experimental procedure. Venn diagram (B) and heat map (C) of genes that were significantly differentially regulated upon FK866 treatment compared with DMSO-treated groups of WT LMO2 or K74/78 LMO2 expressing MOLT4 cells (adjusted value P < .05). Genes known to play a role in tumorigenesis are marked in red. (D) Pathway overrepresentation analysis using a list of significantly regulated genes from panel C revealed top significantly enriched signal transduction pathways that are NAMPT-LMO2 dependent. (E) iRegulon transcription factor motif enrichment analysis using a list of significantly upregulated genes from (C) revealed enrichment of genes (normalized enrichment score >3.0 and area under the curve >0.03) with NAMPT-LMO2–specific transcription factor motifs.
The NAMPT-NAD+-SIRT2 pathway is essential at early-stage hematopoietic differentiation of iPS cells
Previously, we demonstrated that NAMPT activates myeloid differentiation of CD34+ bone marrow-derived HSC.23 LMO2 plays an important role during early stages of hematopoietic differentiation.4,5 Knowing that leukemogenic role of LMO2 in T-ALL may be linked to the abnormal reactivation of the HSC-specific transcriptional program in T-lymphocyte progenitors,16 we aimed to study whether NAMPT-mediated deacetylation of LMO2 is playing a role during early steps of hematopoietic differentiation. We performed a serum- and feeder-free monolayer hematopoietic differentiation of iPS cells51,52 in the presence of FK866, AC93253, or DMSO (Figure 6A). Interestingly, the percentage of KDR+CD34+ hematopoietic progenitor cells on day 6 of culture was noticeably decreased by inhibition of NAMPT or SIRT2 compared with the control DMSO-treated group (Figure 6B-C), whereas the percentage of KDR+CD34− was not affected and the percentage of KDR−CD34− cells was even increased at the highest inhibitor concentration (supplemental Figure 5A-D). Inhibition of SIRT1 had no effect on the formation of hematopoietic cells from iPS cells (supplemental Figure 5E-G). Intracellular NAD+ levels in differentiated iPS cells treated with FK866 were markedly suppressed on day 6 of culture in comparison with the DMSO control group (supplemental Figure 5H). Moreover, the emergence of CD43+ hematopoietic progenitor cells on day 13 of culture as well as colony-forming unit formation was completely suppressed after SIRT2 inhibition (Figure 6D-E). These results indicate that SIRT2 is essential during early hematopoietic differentiation of human iPS cells downstream of the NAMPT-NAD+ signaling pathway.
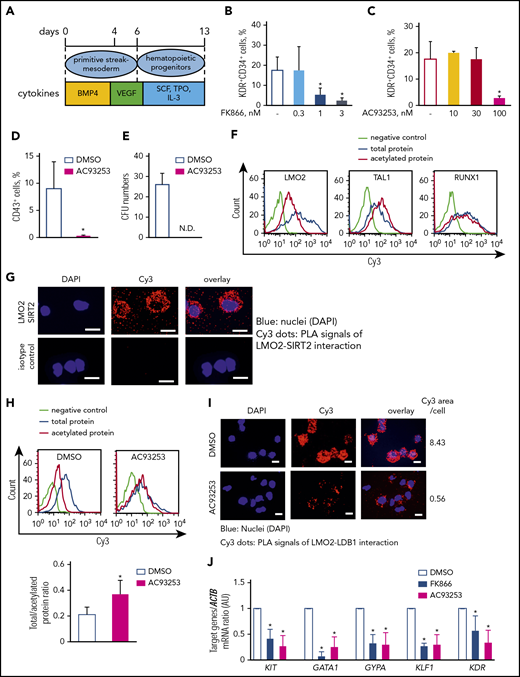
The NAMPT-NAD+-SIRT2 pathway plays an essential role in early-stage hematopoietic differentiation of iPS cells via LMO2 deacetylation. (A) Schematic of the serum- and feeder-free monolayer human iPS cells hematopoietic differentiation system. (B-C) The percentage of KDR+CD34+ cells was evaluated in differentiated iPS cells on day 6 treated with FK866 (B) or AC93253 (C). The same concentration of DMSO was used as a vehicle control. Data show means ± SD and are derived from 3 independent experiments, each in triplicate. *P < .05. (D-E) The percentage of CD43+ cells was evaluated by flow cytometry (D), and colony-forming unit assays were performed in differentiated iPS cells on day 13 treated with 100 nM AC93253 during differentiation (E). The same concentration of DMSO was used as a vehicle control. Data are mean ± SD derived from 3 independent experiments, each in triplicate. *P < .05. (F) Representative histograms of the Duolink-FACS analysis on the acetylation of the indicated proteins in differentiated iPS cells on day 6. Blue, signal from total protein; red, signal from acetylated protein; lime green, negative control. (G) Duolink in situ PLA assay in the differentiated iPS cells on day 6 of culture. Cells were stained with anti-LMO2 and anti-SIRT2 antibodies. Representative images are depicted. Scale bars: 10 μm. (H) Representative histograms of the Duolink-FACS analysis on the acetylation of LMO2 protein in differentiated iPS cells on day 6 in the absence or presence of 100 nM AC93253. The same concentration of DMSO was added as a vehicle control. Blue, signal from total protein; red, signal from acetylated protein; lime green, negative control. Graph bars of the total to acetylated protein Duolink-FACS signal ratio indicates the mean fluorescence intensity (MFI) of acetylated LMO2 protein signal divided by the MFI of total LMO2 protein signal. Data are mean ± SD from 3 independent experiments. *P < .05. (I) Duolink in situ PLA assay in the differentiated iPS cells on day 6 of culture treated with 100 nM AC93253. The same concentration of DMSO was added as a vehicle control. Cells were stained with anti-LDB1 and anti-LMO2 antibodies. Representative images are depicted. Scale bars: 10 μm. (J) mRNA expression levels of the indicated genes in differentiated iPS cells on day 6 of culture in the presence of 3 nM FK866 or 100 nM AC93253 were assessed using quantitative RT-PCR and expressed as AUs. The same concentration of DMSO was added as a vehicle control. Target gene ACTB mRNA expression ratio measured in the control DMSO-treated sample was normalized to 1. Data are mean ± SD from 3 independent experiments, each in triplicate. *P < .05 compared with DMSO-treated cells. ND, not detected.
The NAMPT-NAD+-SIRT2 pathway plays an essential role in early-stage hematopoietic differentiation of iPS cells via LMO2 deacetylation. (A) Schematic of the serum- and feeder-free monolayer human iPS cells hematopoietic differentiation system. (B-C) The percentage of KDR+CD34+ cells was evaluated in differentiated iPS cells on day 6 treated with FK866 (B) or AC93253 (C). The same concentration of DMSO was used as a vehicle control. Data show means ± SD and are derived from 3 independent experiments, each in triplicate. *P < .05. (D-E) The percentage of CD43+ cells was evaluated by flow cytometry (D), and colony-forming unit assays were performed in differentiated iPS cells on day 13 treated with 100 nM AC93253 during differentiation (E). The same concentration of DMSO was used as a vehicle control. Data are mean ± SD derived from 3 independent experiments, each in triplicate. *P < .05. (F) Representative histograms of the Duolink-FACS analysis on the acetylation of the indicated proteins in differentiated iPS cells on day 6. Blue, signal from total protein; red, signal from acetylated protein; lime green, negative control. (G) Duolink in situ PLA assay in the differentiated iPS cells on day 6 of culture. Cells were stained with anti-LMO2 and anti-SIRT2 antibodies. Representative images are depicted. Scale bars: 10 μm. (H) Representative histograms of the Duolink-FACS analysis on the acetylation of LMO2 protein in differentiated iPS cells on day 6 in the absence or presence of 100 nM AC93253. The same concentration of DMSO was added as a vehicle control. Blue, signal from total protein; red, signal from acetylated protein; lime green, negative control. Graph bars of the total to acetylated protein Duolink-FACS signal ratio indicates the mean fluorescence intensity (MFI) of acetylated LMO2 protein signal divided by the MFI of total LMO2 protein signal. Data are mean ± SD from 3 independent experiments. *P < .05. (I) Duolink in situ PLA assay in the differentiated iPS cells on day 6 of culture treated with 100 nM AC93253. The same concentration of DMSO was added as a vehicle control. Cells were stained with anti-LDB1 and anti-LMO2 antibodies. Representative images are depicted. Scale bars: 10 μm. (J) mRNA expression levels of the indicated genes in differentiated iPS cells on day 6 of culture in the presence of 3 nM FK866 or 100 nM AC93253 were assessed using quantitative RT-PCR and expressed as AUs. The same concentration of DMSO was added as a vehicle control. Target gene ACTB mRNA expression ratio measured in the control DMSO-treated sample was normalized to 1. Data are mean ± SD from 3 independent experiments, each in triplicate. *P < .05 compared with DMSO-treated cells. ND, not detected.
LMO2 is deacetylated by NAMPT/SIRT2 during the early stages of hematopoietic differentiation of iPS cells
To identify whether LMO2 is a downstream target of the NAMPT-NAD+-SIRT2 mediated deacetylation at the early stage of hematopoietic differentiation, we compared the acetylation state of LMO2 and other hematopoietic transcription factors essential for early-stage hematopoiesis (TAL1 and RUNX1)5 in iPS-derived hematopoietic cells on day 6 of culture. We used the Duolink-proximity ligation assay (PLA)-FACS,53 which allows identification of endogenous protein complexes or protein modifications in single cells, and quantified the signal using flow cytometry.54 Signal from total protein was obtained by staining with a primary antibody specific for the protein of interest; signal from acetylated protein was obtained using a combination of primary antibody and acetyl-lysine antibody. Interestingly, only LMO2 showed increased deacetylation in iPSCs-derived HSCs on day 6 of culture (Figure 6F; supplemental Figure 6A). The Duolink assay revealed an interaction of LMO2 and SIRT2 proteins in iPSC-derived HSCs on day 6 of culture (Figure 6G) and inhibition of SIRT2 resulted in elevated acetylation of LMO2 as well as diminished interaction with LDB1 compared with control DMSO treated cells (Figure 6H-I; supplemental Figure 6B-C). Target genes of the TAL1 complex as well as FOS and FRZB (early hematopoiesis genes) mRNA expression were also suppressed in iPS cell-derived HSCs on day 6 of culture in the presence of NAMPT- or SIRT2-specific inhibitors (Figure 6J; supplemental Figure 6D). These data suggest an essential role of NAMPT-NAD+-SIRT2-mediated deacetylation of LMO2 for the activation of TAL1 complexes in early hematopoiesis.
Inhibition of the NAMPT-NAD+-SIRT2 pathway with morpholinos abrogates early hematopoiesis in zebrafish embryos
We further evaluated the role of the NAMPT-NAD+-SIRT2 pathway during early hematopoiesis in vivo in zebrafish. In zebrafish embryos, developing blood cells appear in the blood island located just before the yolk extension around 24 hpf.55 To analyze early hematopoiesis, we used transgenic zebrafish Tg(-6.35drl:EGFP) expressing GFP under the control of the draculin promoter, which is expressed in hematopoietic progenitor cells within the lateral plate mesoderm.36 We knocked down zebrafish orthologs for NAMPT and SIRT2, (nampta and sirt2) by injecting 1-cell-stage zebrafish embryos with antisense MOs.37 MO-induced mRNA knockdown was efficient (supplemental Figure 7A). Although nampta- or sirt2-knockdown embryos showed tail defects, these phenotypes could be rescued by coinjection with expression plasmid encoding human NAMPT or SIRT2, indicating nampta and sirt2 specific knockdown (supplemental Figure 7B). We quantified early hematopoiesis in zebrafish embryos by counting GFP+ cells in the developing blood island at 24 hpf and found that nampta and sirt2 MO-injected embryos had markedly reduced numbers of hematopoietic cells compared with uninjected control embryos (Figure 7A), which could be rescued by coinjection with expression plasmid of human NAMPT or SIRT2 (supplemental Figure 7C-D). In line with impeded hematopoiesis 24 hpf, NAD+ levels as well as mRNA expression levels of downstream target genes of the TAL1 complex were downregulated by knockdown of nampta and sirt2 genes (Figure 7B; supplemental Figure 7E). These data suggest that the NAMPT-NAD+-SIRT2 pathway is indispensable for early hematopoiesis, most likely through the activation of the TAL1 complex in zebrafish embryos.
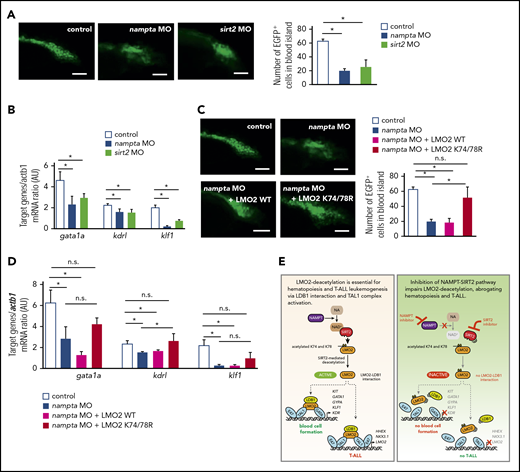
Morpholino-mediated inhibition of the NAMPT-NAD+-SIRT2 pathway impedes early hematopoiesis in zebrafish embryos. (A,C) Quantification of EGFP+ hematopoietic cells in uninjected control and morpholino (MO)/plasmid injected Tg(-6.35drl:EGFP) zebrafish embryos at 24 hpf. Representative images and numbers of EGFP+ hematopoietic cells in the blood islands are shown. Data show mean ± SD (n = 5). *P < .05. Scale bars: 50 μm. (B,D) mRNA expression levels of the indicated genes in control and MO/plasmid injected zebrafish embryos at 24 hpf were assessed using quantitative RT-PCR. Target gene/actb1 mRNA expression ratios are shown. Data show mean ± SD from 3 independent experiments, each in triplicate. *P < .05. (E) Schematic for the NAMPT/SIRT2-mediated lysine deacetylation of the LMO2 transcription factor that is essential for the interaction of LMO2 with LDB1 and formation of the TAL1 complex. This activates the oncogenic transcriptional program and promotes T-cell leukemia development or induces blood cell formation.
Morpholino-mediated inhibition of the NAMPT-NAD+-SIRT2 pathway impedes early hematopoiesis in zebrafish embryos. (A,C) Quantification of EGFP+ hematopoietic cells in uninjected control and morpholino (MO)/plasmid injected Tg(-6.35drl:EGFP) zebrafish embryos at 24 hpf. Representative images and numbers of EGFP+ hematopoietic cells in the blood islands are shown. Data show mean ± SD (n = 5). *P < .05. Scale bars: 50 μm. (B,D) mRNA expression levels of the indicated genes in control and MO/plasmid injected zebrafish embryos at 24 hpf were assessed using quantitative RT-PCR. Target gene/actb1 mRNA expression ratios are shown. Data show mean ± SD from 3 independent experiments, each in triplicate. *P < .05. (E) Schematic for the NAMPT/SIRT2-mediated lysine deacetylation of the LMO2 transcription factor that is essential for the interaction of LMO2 with LDB1 and formation of the TAL1 complex. This activates the oncogenic transcriptional program and promotes T-cell leukemia development or induces blood cell formation.
While the morpholino knockdown data has to be confirmed with genetic mutants, injection of zebrafish embryos with expression plasmid of human deacetylation-mimic LMO2 mutant (K74/78R), but not WT LMO2, rescued nampta MO-induced defective hematopoiesis and mRNA expression of TAL1 complex target genes at 24 hpf (Figure 7C-D). Together with our results in human iPS cells, these results indicate that deacetylation of LMO2 at K74/78 by NAMPT and SIRT2 is crucial for early vertebrate hematopoiesis.
Discussion
In the present study, we demonstrated for the first time that NAMPT and SIRT2 are essential for the proliferation and survival of T-ALL blasts in vitro and in vivo. We also found that NAMPT and SIRT2 are important for blood cell formation in an iPS cell model in vitro and in morpholino-mediated knockdown experiments in zebrafish embryos in vivo. Therefore, we have opened a new line of investigation into the role of NAMPT-mediated protein deacetylation in developmental hematopoiesis and T-ALL leukemogenesis. It would be interesting to investigate whether NAMPT/SIRT2-mediated protein deacetylation governs the development of other tissues and organs of, for example, mesoderm origin, and whether SIRT2 or other SIRTs are involved.
The dose- and cell type-dependence of NAMPT in the hematopoietic system is well documented. Thus, hyperactivated NAMPT and SIRT2 have been shown to induce uncontrolled proliferation of hematopoietic progenitors and cause acute myeloid leukemia.24 We demonstrated that inhibition of NAMPT or SIRT2 suppressed proliferation and induced apoptosis of T-ALL cells in vitro and in vivo. We previously published evidence for an important role of NAMPT in later stages of the myeloid specification of hematopoietic progenitors that depend on granulocyte colony-stimulating factor or granulocyte-macrophage colony-stimulating factor.23,27 Most likely, NAMPT is also activated by other growth factors essential for the induction of embryonic hematopoiesis, such as BMP4, VEGF, and basic fibroblast growth factor, or the development of T-ALL, such as common γ-chain signaling cytokines (gamma c-cytokines), including IL-2, IL-4, IL-7, and IL-9.56,-58 We recently determined the involvement of the IL-13 pathway in NAMPT-triggered myeloid differentiation.26 Less is known about the role of NAMPT and SIRTs in T lymphopoiesis. Bruzzone et al showed impaired proliferation and function of activating T lymphocytes upon NAMPT depletion in experimental autoimmune encephalomyelitis.59 SIRT1 plays a role in Foxp3+ T-regulatory cell function60 and in human CD8+ memory T cells.61 Interestingly, we found that SIRT2, but not SIRT1, is important during early stages of blood cell formation and T-ALL development downstream of NAMPT activation. SIRT1 and SIRT2 have a high degree of structural similarity and may have similar functions and targets. However, there are also reports that SIRT1 possesses specific functions that are not shared by SIRT2. The same is true for SIRT2.27,-29 In our system, it is obvious that these 2 SIRTs exhibit different functions and have different downstream effectors. Previous investigations by Han et al and Ou et al demonstrated that a SIRT1 deficiency causes increased apoptosis and diminished hematopoietic differentiation in mouse ES cells.31,62 In contrast, we found no effect of SIRT1 inhibition on early hematopoietic differentiation, a difference that may be explained by the differences in species and/or cell type used. Whether SIRT2 also plays a role in apoptosis induction and/or hematopoietic differentiation of mouse ES cells remains to be investigated.
Importantly, we indicated for the first time that LMO2 is a downstream target of NAMPT signaling, showing that NAMPT/SIRT2-mediated LMO2 activation by deacetylation on lysines K74 and K78 is essential for LMO2 interaction with LDB1 and formation of a transcriptionally active TAL1 complex (Figure 7E). We found that LMO2 is deacetylated by NAMPT/SIRT2 during early stages of hematopoietic differentiation and T-ALL development (Figure 7E). This is the first report describing activation of LMO2, a developmentally important protein, by deacetylation on K74 and K78 within the LIM1 domain, which is essential for the association of LMO2 with LDB1 protein and TAL1 complex activation. Knowing this, it has become possible to perform structure-based screens of chemical compounds to identify candidates that either prevent LMO2 deacetylation and LDB1 binding in T-ALL, or induce this interaction and enhance blood cell formation. Holen et al63 and von Heideman et al64 already described FK866’s toxicity and safety analyzing the first clinical studies for FK866 treatment in cancer patients. The dose-limiting toxicity of FK866 is thrombocytopenia and various gastrointestinal symptoms including nausea, vomiting and diarrhea. In our study, daily treatment of NSG mice with FK866 for up to 21 days was well tolerated with no premature deaths, as already described by Nahimana et al65 and Matheny et al.66 Only some minor side effects were observed, such as mild fatigue and reduced movement compulsion. In zebrafish embryos, FK866 treatment did not show any severe side effects even if used in higher concentrations.
LMO2 is expressed in early T-cell progenitors and is normally switched off during T-lymphocyte differentiation.11,,,-15 In T-ALL, LMO2 continues to be expressed owing to recurrent chromosomal translocations, deletions in an upstream negative regulatory region, somatic acquisition of a neomorphic promoter, or gene therapy-related retroviral insertional mutagenesis at the LMO2 locus.8,22,67,,-70 In our T-ALL patient cohort, we did not compare LMO2 expression levels to that of early thymocyte progenitors, but the samples selected for further analysis showed comparable LMO2 expression level to the LMO2 dependent MOLT4 T-ALL cell line. Activated LMO2 reactivates an HSC-specific transcriptional program in T-cell progenitors, leading to enhanced self-renewal and proliferation, with reduced capacity for T-cell differentiation and transformation into T-ALL blasts.15,16 These data are in agreement with our observation that LMO2 activation by NAMPT/SIRT2-mediated lysine deacetylation is essential for both formation of HSCs and T-ALL leukemogenesis. Most likely, deacetylated LMO2 activates common downstream targets that are important a specific transcriptional program in HSCs and transformation of T-ALL blasts. At the same time, we cannot exclude the involvement of other transcriptional targets in addition to LMO2 that are activated by NAMPT and SIRTs in HSCs and/or T-ALL cells. In addition, NAMPT-independent mechanisms of LMO2 activation in HSCs and T-ALL cells should be considered.
Using RNA-Seq analysis of MOLT4 cells stably expressing WT or deacetylated LMO2 mutant, we were able to identify signaling pathways such as cytochrome P450, tryptophan degradation, biological oxidation, P53- or TNF-α downstream signaling cascades to be regulated by NAMPT-mediated LMO2 deacetylation. Components of the cytochrome pathway, CYPE1 and CYP1B1, that were downregulated upon NAMPT inhibition in a LMO2-dependent manner, are known to play a role in the ALL pathogenesis.71,-73 Additionally, tryptophan degradation may play a role in the tumorigenesis and may be evaluated further for the potential drug targeting.74,75 Most probably, LMO2 is involved in the metabolic reprogramming of HSCs during T-leukemogenesis.76,-78 P53 mutations are frequent finding in ALL that correlates with poor chemotherapy response.79 TNF-α is known to be involved in different steps of leukemogenesis, including cellular transformation, proliferation, survival, and resistance to chemotherapy.80,,-83 In iRegulon analysis, we identified target gene enrichment for known T-ALL-associated transcription factors, such as PITX,84 ZNF695,85 MEF2 family,86,,,,-91 and EP300.92 These data further confirmed the important role of the LMO2 activation by NAMPT-mediated deacetylation in the T-ALL development. It would be interesting to investigate functional connection between identified candidate genes and pathways and NAMPT-activated LMO2 in the pathogenesis of T-ALL and early hematopoiesis.
The 2 experimental models for studying early steps in hematopoiesis used here (in vitro iPS cells and in vivo zebrafish) have offered us an excellent opportunity to study the involvement of NAMPT/SIRTs in differentiation processes of diverse layers/organs and at defined developmental stages. Our observations may help establish improved culture conditions for modulating NAMPT/SIRT signaling (eg, for large-scale in vitro generation of hematopoietic cells from iPS cells).
Taken together, our findings demonstrate that a novel posttranslational mechanism, NAMPT/SIRT2-mediated lysine deacetylation, is involved in regulating the developmentally important protein LMO2. This mechanism could prove profoundly important for developmental biologists, tumor biologists, and clinical scientists/oncologists. Our observations have translational significance, suggesting the possibility of clinically targeting T-ALL leukemia cells through specific inhibition of NAMPT-mediated LMO2 deacetylation. They also have implications for the large-scale in vitroproduction of hematopoietic cells through modulation of the NAMPT/SIRT2/LMO2 axis.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Nico Lachmann and Thomas Moritz (Institute of Experimental Hematology, Hannover Medical School) for providing the human iPS cell line hCD34-iPSC16, Qiang Tong (Baylor College of Medicine) for providing pcDNA3.1-SIRT2-flag plasmid, Igor Slukvin (University of Wisconsin School of Medicine and Public Health) for providing pSIN4-EF1a-LMO2-IRES-Puro and pSIN4-EF1a-GATA1-IRES-Puro plasmids, and Robert Benezra (Memorial Sloan Kettering Cancer Center) for providing pcDNA3-hE47 plasmid. Cell sorting and flow cytometry sample acquisition was done on shared instruments of the Flow Cytometry Core Facility Tübingen.
This work was supported by the Applied Clinical Research program from the Faculty of Medicine of the Tübingen University (T.M.); the Fortüne program from the Faculty of Medicine, the Tübingen University (T.M.); the Research Fellowship of Uehara Memorial Foundation (T.M.); Deutsche Forschungsgemeinschaft (DFG) (J.S., B.B., and N.A.); Sander Stiftung (A.K.); and Schickedanz Kinderkrebsstiftung (J.S.).
Authorship
Contribution: T.M., J.S., and K.W. made initial observations, designed the experiments, analyzed the data, supervised experimentation, and wrote the manuscript; T.M. performed the main experiments, including zebrafish knockdown; A.-C.K., M.R., S.S., and M.A. designed and performed mouse transplantation experiments; T.M. and M.K. performed Duolink experiments; T.M., Y.X., C.L., B.O., and U.H.-K. performed western blots and immunoprecipitation; T.M. and B.F. made reporter gene assays; R.B. cultured human iPS cells; R.B. and K.H. performed quantitative polymerase chain reaction experiments; Y.X. and M.N. performed CRISPR/Cas9-mediated SIRT2 gene knockout; M.D.H. conducted structural analysis of LMO2; N.C. performed RNA-sequencing; M.N. analyzed RNA-sequencing data; P.M. designed and supervised the zebrafish experiments; B.B. and N.A. performed the zebrafish xenograft experiments and analyzed the data; M.E. and R.H. provided T-ALL patients material; and C.J.G. assisted with the interpretation of data and provided insightful comments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for T.M. is International Research Center for Medical Sciences, Kumamoto University, Kumamoto, Japan.
Correspondence: Julia Skokowa, Department of Hematology, Oncology, Clinical Immunology and Rheumatology, University Hospital Tübingen, Otfried-Mueller Str 10, 72076 Tübingen, Germany; e-mail: julia.skokowa@med.uni-tuebingen.de; and Tatsuya Morishima, International Research Center for Medical Sciences, Kumamoto University, 2-2-1 Honjo, Chuo-ku, Kumamoto City 860-0811, Japan; e-mail: tatsuyam@kumamoto-u.ac.jp.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal