

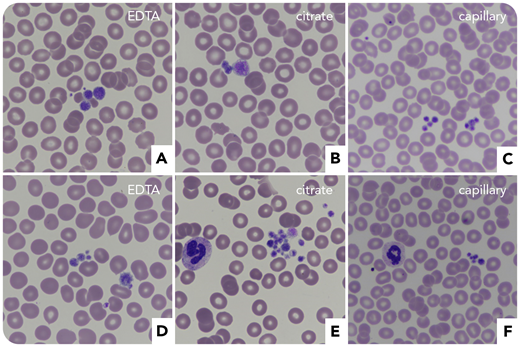
A 63-year-old woman with a history of immune thrombocytopenic purpura (ITP) was referred for subarachnoid hemorrhage and subdural hematoma after a fall. At admission, she had 24 × 109/L platelets with aggregates on blood smears (EDTA, citrate, and capillary) and large platelets (panels A-C, May-Grünwald Giemsa stain, original magnification ×60). IV immunoglobulins (IVIGs) and corticosteroids were ineffective, while platelet concentrates showed a transient efficacy. Because of the ITP, eltrombopag was prescribed, which normalized platelet counts and stopped the hemorrhage. Incidentally, her sister revealed that she also was suspected of ITP and that there was a notion of von Willebrand disease (VWD) in the family. She was thus also tested, which disclosed severe thrombocytopenia (48 × 109/L platelets) with aggregates (panels D-F, May-Grünwald Giemsa stain, original magnification ×60), factor VIII:C 1.02 IU/mL, VWF:ristocetin cofactor 0.4 IU/mL, and VWF:antigen 0.8 IU/mL (ratio VWF:ristocetin cofactor/VWF:antigen, 0.5).
Both sisters were then diagnosed with VWD type 2B (“Montreal disease”), and next-generation sequencing of VWF gene exon 28 revealed the c.3946G>A (p.V1316M) mutation. The patient’s history showed that this could have been suspected earlier if the family context was investigated. Indeed, she had presented a nontraumatic hemoperitoneum requiring splenectomy and platelet transfusion. Significant bleeding after her first pregnancy had led to inefficient corticosteroids and IVIG during further deliveries, with platelet transfusions being necessary following childbirth.
A 63-year-old woman with a history of immune thrombocytopenic purpura (ITP) was referred for subarachnoid hemorrhage and subdural hematoma after a fall. At admission, she had 24 × 109/L platelets with aggregates on blood smears (EDTA, citrate, and capillary) and large platelets (panels A-C, May-Grünwald Giemsa stain, original magnification ×60). IV immunoglobulins (IVIGs) and corticosteroids were ineffective, while platelet concentrates showed a transient efficacy. Because of the ITP, eltrombopag was prescribed, which normalized platelet counts and stopped the hemorrhage. Incidentally, her sister revealed that she also was suspected of ITP and that there was a notion of von Willebrand disease (VWD) in the family. She was thus also tested, which disclosed severe thrombocytopenia (48 × 109/L platelets) with aggregates (panels D-F, May-Grünwald Giemsa stain, original magnification ×60), factor VIII:C 1.02 IU/mL, VWF:ristocetin cofactor 0.4 IU/mL, and VWF:antigen 0.8 IU/mL (ratio VWF:ristocetin cofactor/VWF:antigen, 0.5).
Both sisters were then diagnosed with VWD type 2B (“Montreal disease”), and next-generation sequencing of VWF gene exon 28 revealed the c.3946G>A (p.V1316M) mutation. The patient’s history showed that this could have been suspected earlier if the family context was investigated. Indeed, she had presented a nontraumatic hemoperitoneum requiring splenectomy and platelet transfusion. Significant bleeding after her first pregnancy had led to inefficient corticosteroids and IVIG during further deliveries, with platelet transfusions being necessary following childbirth.
For additional images, visit the ASH Image Bank, a reference and teaching tool that is continually updated with new atlas and case study images. For more information, visit http://imagebank.hematology.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal