

In this issue of Blood, demonstrate the pleiotropic immunomodulatory effects by which the Janus kinase (JAK) inhibitor ruxolitinib ameliorates hypercytokinemia and organ injury in 2 distinct murine models of hemophagocytic lymphohistiocytosis (HLH).1 By demonstrating both interferon-γ (IFN-γ)–dependent and IFN-γ–independent effects, the authors provide compelling preclinical data supporting the potential superiority of ruxolitinib when compared with IFN-γ inhibition alone. In doing so, the authors also identify a previously underrecognized role of neutrophil cytotoxicity in the pathobiology of HLH-induced multiorgan dysfunction.
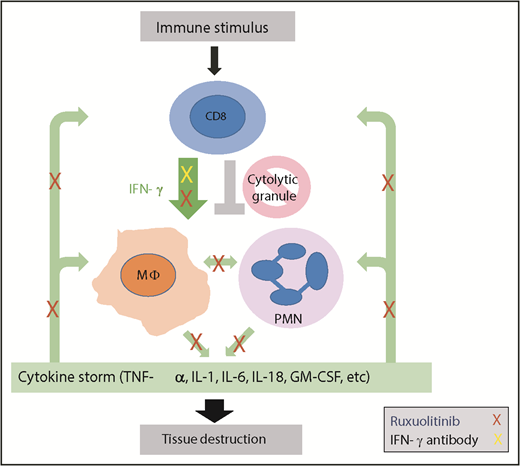
HLH is a hyperinflammatory disease triggered by activating stimuli, including malignancy, infection, or autoimmunity, that leads to CD8+ T-cell activation, IFN-γ production, and activation of immune effector cells that produce additional cytokines. Due to inherited or acquired defects in cytolytic granule release, termination of the immune response does not occur. Continuous cytokine production ensues (green arrows), leading to a cytokine storm with continued immune effector cell activation and downstream tissue damage. The work by Albeituni et al implicates neutrophils as an additional effector of this tissue damage. IFN-γ blockade (yellow X) specifically blocks the driving cytokine in this process.9 The JAK inhibitor ruxolitinib (red Xs) blocks IFN-γ, but also has more pleiotropic effects impacting additional cytokine-mediated signals, including those that impact deleterious neutrophil activity. MΦ, macrophage; PMN, polymorphonuclear neutrophil.
HLH is a hyperinflammatory disease triggered by activating stimuli, including malignancy, infection, or autoimmunity, that leads to CD8+ T-cell activation, IFN-γ production, and activation of immune effector cells that produce additional cytokines. Due to inherited or acquired defects in cytolytic granule release, termination of the immune response does not occur. Continuous cytokine production ensues (green arrows), leading to a cytokine storm with continued immune effector cell activation and downstream tissue damage. The work by Albeituni et al implicates neutrophils as an additional effector of this tissue damage. IFN-γ blockade (yellow X) specifically blocks the driving cytokine in this process.9 The JAK inhibitor ruxolitinib (red Xs) blocks IFN-γ, but also has more pleiotropic effects impacting additional cytokine-mediated signals, including those that impact deleterious neutrophil activity. MΦ, macrophage; PMN, polymorphonuclear neutrophil.
HLH is a hyperinflammatory disorder characterized by massive immune cell activation leading to severe multiorgan injury that culminates in mortality in up to 50% of affected children and adults.2,3 Immune cell activation can be triggered by infection, malignancy, or autoimmunity and is propagated by primary or acquired defects in T-cell and natural killer (NK)–cell cytotoxicity that preclude their ability to terminate the immune response. Thus, hypercytokinemia is both a result and driver of immune cell activation. The cytokine storm of HLH involves elevations in IFN-γ, tumor necrosis factor-α (TNF-α), and interleukin-1 (IL-1), IL-2, IL-6, and IL-18. While CD8+ T lymphocytes and macrophages are known to propagate HLH-related inflammation, the role of other cellular lineages that may be activated in the wake of this cytokine storm and that contribute to end-organ damage remains unclear. Most patients are treated based on the HLH 94 and 2004 protocols with dexamethasone and etoposide. While efficacious in a subset of patients, systemic toxicities are signficant.4,5
The hypercytokinemia of HLH has become an attractive alternative therapeutic target. Compelling preclinical and clinical data led to the recent US Food and Drug Administration approval of the IFN-γ–blocking antibody emapalumab (Gamifant) for patients with relapsed, refractory, or progressive primary HLH (NCT01818492). However, a potential limitation of targeting a single cytokine is that it does not impact the source of cytokine production or other cytokines that may propagate immune activation and end-organ damage. Several case reports and preclinical models have highlighted the JAK inhibitor ruxolitinib as an alternative approach. JAK inhibition represents an appealing target for the treatment of hypercytokinemia, as JAKs are expressed on numerous immune cell lineages, including myeloid, lymphoid, dendritic, and CD56+ NK cells, and mediate signaling of multiple cytokines, including IFN-α, IFN-β, IFN-γ, IL-2, IL-3, IL-4, IL-6, IL-7, IL-9, IL-12, IL-10, IL-15, IL-21, granulocyte macrophage colony-stimulating factor (GM-CSF), and granulocyte colony-stimulating factor.6 However, while inhibition of multiple cytokines could be theoretically more beneficial, increased toxicity is also a possibility. Studies comparing these strategies are lacking.
Albeituni et al address this issue by evaluating whether JAK1/2 inhibition to block signaling of multiple cytokines is superior to single-cytokine blockade of IFN-γ. In a model of primary HLH (lymphocytic choriomeningitis virus [LCMV] infection in perforin-deficient mice), JAK1/2 inhibition led to less thrombocytopenia, less hepatosplenomegaly with less CD8+ T-cell/monocyte/neutrophil infiltration and activation, and lower cytokine levels (IL-6, TNF-α, GM-CSF, MCP-1, and MIP-1α) relative to IFN-γ inhibition alone. Further, treatment with ruxolitinib on days 4 to 9 after disease induction was associated with significantly greater 35-day survival than IFN-γ inhibition alone (14/14 vs 5/13 mice). In a model of secondary HLH (TLR9 stimulation with IL-10 receptor blockade), JAK1/2 inhibition was also superior to IFN-γ inhibition.
This work highlights 2 important findings that merit discussion. First, the use of JAK1/2 inhibition was clearly superior to IFN-γ inhibition in reducing hypercytokinemia, immune activation, organomegaly, and death in these murine models of HLH. Teleologically, this is perhaps not surprising in that an anti-IFN-γ antibody can serve as a sponge to decrease the amount of deleterious cytokine present while JAK1/2 inhibition is akin to turning off the faucet and blocking the effects of multiple cytokines (see figure). Second, Albeituni et al identified a critical role of neutrophil-mediated tissue injury in the pathobiology of HLH. Specifically, the authors noted that splenic neutrophils from LCMV-infected perforin-deficient mice upregulated triggering receptor expressed on myeloid cells-1 (TREM-1) and increased production of intracellular TNF-α, MIP-1α, MIP-1β, and IL-1β. This endotype was ameliorated with JAK1/2 inhibition, but not IFN-γ inhibition. Interestingly, the poorer survival of mice treated with IFN-γ inhibition was rescued by the addition of neutrophil-depleting antibodies, but not anti-IL-6 or anti–TNF-α antibodies. This supports a role for neutrophil activation in the pathobiology of HLH and highlights possible similarities between HLH-mediated organ injury and severe organ injury seen in other critical illnesses such as sepsis and acute respiratory distress syndrome (ARDS).7,8 For example, in ARDS, activated alveolar macrophages recruit neutrophils by releasing GM-CSF, MCP-1, IL-1α, IL-8, and TNF-α (cytokines also elevated in HLH). Neutrophils then act as the primary effectors of tissue injury by releasing proteases, reactive oxygen species, and neutrophil extracellular traps. These findings suggest that targeting the myeloid compartment may have therapeutic benefit in HLH. However, the potential benefits of neutrophil inhibition in the acute stages of HLH will need to be balanced with the potential infectious consequences of long-term impairment of neutrophil function.
Ultimately, the key to improving outcomes for patients with HLH likely depends on both mitigating immune cell activation and counteracting the mechanisms by which immune cell activation directly propagates fatal organ injury. While the work by Albeituni et al does not negate the benefits of blocking IFN-γ, which is a clearly established driver of HLH,9 it does suggest that some patients may receive more benefit from ruxolitinib than from single-cytokine targeted therapy. Nonetheless, caution is warranted. Although ruxolitinib has been safely used for multiple diseases and in the context of combination chemotherapy,6 the safety of blocking multiple cytokines in the context of a critically ill patient with HLH needs further evaluation. Future clinical trials are clearly indicated.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal