

Altered metabolism has long been recognized as a hallmark of cancer,1 but recently, the direct effect of metabolites on epigenetic regulation has become more appreciated. In this issue of Blood, 2 use a mouse model of acute myeloid leukemia (AML) to show that the metabolic regulator adenosine monophosphate–activated protein kinase (AMPK) controls acetyl-coenzyme A (acetyl-CoA) homeostasis.
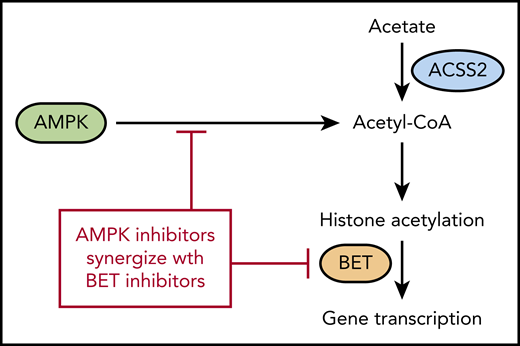
AMPK regulates acetyl-CoA levels, which reduces histone acetylation and makes cells more sensitive to BET inhibitors. ACSS2 also contributes to acetyl-CoA levels by converting free acetate to acetyl-CoA.
AMPK regulates acetyl-CoA levels, which reduces histone acetylation and makes cells more sensitive to BET inhibitors. ACSS2 also contributes to acetyl-CoA levels by converting free acetate to acetyl-CoA.
Warburg’s3 observation that cancers consume large amounts of glucose despite available oxygen led to his hypothesis that aerobic glycolysis was a hallmark of cancer growth. Although glycolysis is frequently observed in cancer, most cancer cells still use mitochondrial oxidative phosphorylation and the citric acid cycle to generate energy and substrates for cell growth. Many of the downstream metabolites of glucose metabolism were later found to be important as cofactors for proteins that regulate gene transcription, thus providing a direct link between metabolism and regulation of gene expression. Two examples of this are α-ketoglutarate and acetyl-CoA.
α-Ketoglutarate is produced by isocitrate dehydrogenase (IDH) and is an important cofactor for several enzymes, including TET2, which is involved in demethylation of cytosine in DNA. TET2 itself is frequently mutated in AML, but TET2 can also be functionally inactivated by IDH1 and IDH2 mutations. Mutated IDH leads to the production of the oncometabolite 2-hydroxyglutarate instead of α-ketoglutarate, and 2-hydroxyglutarate inhibits the demethylase activity of TET2. IDH1 and IDH2 inhibitors restore TET2 activity, gene transcription, and cause differentiation of leukemia cells. IDH1 and IDH2 inhibitors were recently approved by the US Food and Drug Administration for AML, which proves that altered cancer metabolism and gene transcription can be effectively targeted.4
Acetyl-CoA is another product of the citric acid cycle and is the building block for fatty acid synthesis as well as a cofactor for acetylation of proteins. Although cancer cells are often thought to be dependent upon glycolysis, they can readily adapt to use energy sources other than glycolysis during periods of nutrient deprivation. AMPK activates several catabolic pathways to increase acetyl-CoA in addition to increasing glucose transport into the cell.5 A previous article by the authors demonstrated that genetic deletion of AMPK in the MLL-AF9 mouse model of AML impairs glucose metabolism, which leads to reduced growth and delayed leukemogenesis.6 In the Jiang et al article in this issue, the authors investigated the effect of AMPK deletion on metabolites other than glucose. As expected, numerous glucose metabolites were decreased after AMPK deletion. The authors then performed a rescue screen with the metabolites and found that acetate was most effective at restoring growth. Acetate was also able to inhibit differentiation and increased expression of the genes Myc, Meis1, and HoxA9, suggesting that it was playing a role beyond energy metabolism. Because acetate is converted into acetyl-CoA by the acyl-CoA synthetase short chain (ACSS) family of proteins, the authors used CRISPR-Cas to verify that the Acss2 gene is required for converting acetate back into acetyl-CoA (see figure). Accordingly, Acss2 deletion also delayed leukemogenesis and prolonged survival in the MLL-AF9 leukemia model, similar to deletion of AMPK.
The authors then looked at the histones in the AMPK-deleted leukemia cells and discovered that acetylation is reduced on H3 and H4 histones. Acetylated histones are recognized by bromodomain and extra-terminal domain (BET) family of proteins, and AMPK-deleted cells were found to have decreased expression of several genes regulated by BET proteins. This suggested that inhibition of AMPK and BET may be synergistic because they regulate many of the same genes. The authors then confirmed this synergy by using both genetic deletion and small-molecule inhibitors of AMPK and BET proteins.
As noted by the authors, the link between metabolism, acetyl-CoA, and histone acetylation has been previously reported,7 but the synergy between histone acetylation and bromodomain inhibitors in the MLL-AF9 mouse model is worth noting. Although there are reports of BET inhibitors having activity in leukemia, the Jiang et al study suggests that multiple agents targeting the same pathway may be more effective. That being said, there are still many questions about how to translate these findings.
Because both AMPK and BET proteins are present in normal cells, it is difficult to predict the adverse effects of AMPK and BET inhibition. The authors created an AMPK/Brd4 heterozygous mutant mouse for their experiments that did not have any hematopoietic deficits. Although this suggests that there might be a potential therapeutic window, it remains to be proven in patients.
Another problem with targeting AMPK is that acetate can circumvent decreased acetyl-CoA production, as the authors themselves show in their rescue experiments. Acetate is present in large amounts in the plasma, so it is unclear how effective AMPK inhibition alone will be. Targeting ACSS2 to prevent use of acetate may be a way to avoid this problem. The authors demonstrated that deletion of Acss2 itself slowed leukemia growth in the MLL-AF9 model, and Acss2 deletion has been shown to slow tumor growth in other cancer models.8,9
Drugs that target metabolic pathways are already widely used in hematologic malignancies and include methotrexate, asparaginase, and IDH inhibitors. It should be noted that all of these drugs are most effective when used in combination with other agents. Although the complexity of metabolism and epigenetics is challenging,10 discovering the unique ways that cancer cells adapt to metabolic stress to sustain growth will help guide rational combinations or sequencing of drugs that can enhance response to therapy.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal