

Key Points
The high-resolution structure and dynamics of Ixolaris are presented.
The noncanonical mechanism of factor X inhibition through an allosteric switch at the catalytic site is proposed.

Abstract
Ixolaris is a potent tick salivary anticoagulant that binds coagulation factor Xa (FXa) and zymogen FX, with formation of a quaternary tissue factor (TF)/FVIIa/ FX(a)/Ixolaris inhibitory complex. Ixolaris blocks TF-induced coagulation and PAR2 signaling and prevents thrombosis, tumor growth, and immune activation. We present a high-resolution structure and dynamics of Ixolaris and describe the structural basis for recognition of FX. Ixolaris consists of 2 Kunitz domains (K1 and K2) in which K2 is strikingly dynamic and encompasses several residues involved in FX binding. This indicates that the backbone plasticity of K2 is critical for Ixolaris biological activity. Notably, a nuclear magnetic resonance–derived model reveals a mechanism for an electrostatically guided, high-affinity interaction between Ixolaris and FX heparin-binding (pro)exosite, resulting in an allosteric switch in the catalytic site. This is the first report revealing the structure-function relationship of an anticoagulant targeting a zymogen serving as a scaffold for TF inhibition.
Introduction
Hemostasis is initiated on vessel wall damage, which exposes tissue factor (TF) that activates blood coagulation and collagen that induces platelet aggregation.1 Mechanistically, TF binds FVIIa with formation of catalytically active TF/FVIIa complex, which converts FIX to FIXa, or FX to FXa.2,3 As a result of limited proteolysis reactions that occur at the cell membrane in the presence of cofactors, the coagulation cascade is markedly amplified, leading to intense thrombin generation and clot formation. The TF/FVIIa activity is under control of TF pathway inhibitor (TFPI), a potent physiological anticoagulant comprising 3 tandem Kunitz-type inhibitor domains designated K1-K3. TFPI has a peculiar mechanism of action in which the K2 interacts with the active site of FXa, which works as a scaffold for inhibition of the FVIIa/TF complex. The K1 assists in complex formation through binding to FVIIa catalytic site.2,3
Several studies indicate that the interaction of TFPI with FXa involves a number of other protein-protein interactions, in addition to the active site. The affinity of the separate K2 domain for FXa is about 3 orders of magnitude lower than that of TFPI.4,5 In addition, cleavage of TFPI between K1 and K2 dramatically reduces the affinity of TFPI for FXa.6 Studies employing synthetic peptides or monoclonal antibodies have demonstrated that, in addition to K2, a number of residues in the K1 domain mediate the interaction of the TFPI with FXa.7,8 Thus, it is proposed that K1 provides a high-affinity binding region for FXa that allows subsequent inhibition of the coagulation enzyme by the K2 domain. These results highlight the complexity of the reactions involving TFPI and emphasize the critical role of scaffold FXa in this process. Therefore, understanding the underlying molecular mechanisms of TFPI and related inhibitors may provide the rationale for designing anticoagulants targeting TF in a number of diseases, without major impairment of hemostasis.9-11
The TF pathway plays important roles in normal hemostasis, cardiovascular disease, and thrombosis. Coagulation activation by TF is implicated in cancer progression, cancer-associated thrombosis, and metastasis, although the molecular mechanism is not completely understood.12,13 Overexpression of TF enhances tumor growth, whereas knockdown attenuates tumor expansion.14-16
In this respect, exogenous secretion such as saliva from blood-sucking arthropods contains an immense repertoire of molecules affecting vascular biology.17,18 Ixolaris is a tick salivary anticoagulant containing 2 Kunitz-like domains that binds FXa or FX, which serve as scaffolds for inhibition of TF/FVIIa complex.19 In contrast to TFPI, Ixolaris does not interact with the active site cleft of FXa.19 The canonical mechanism of Kunitz inhibition, as observed for TFPI, resembles the formation of an enzyme-substrate complex and no interaction with the zymogen. In contrast, Ixolaris interacts with similar high affinity to FX and FXa20,21 through (pro)exosites,22 as scaffolds to inhibit TF-FVIIa complex (Kd, 0.67 and 0.25 nM, respectively).19
Additional studies revealed that Ixolaris prevents PAR2 cleavage specifically by human TF signaling complexes, indicating that it may also attenuate inflammatory tonus associated with TF expression.23 Furthermore, in vivo experiments showed that Ixolaris prevents thrombosis in rats with a long half-life,24 and blocks angiogenesis and tumor progression in a glioblastoma model.25 More recently, treatment of chronically infected monkeys with simian immunodeficiency virus with Ixolaris was accompanied by inhibition of monocyte TF activity and a significant decrease in d-dimer and immune activation.26 Altogether, these results suggest that Ixolaris represents a novel therapeutic option to attenuate coagulation and inflammation associated with thrombosis, cancer, and chronically infected patients with HIV.
In the present study, we describe the interaction of Ixolaris with FX. We show that Ixolaris employs both K1 and K2 domains to recognize the heparin-binding (pro)exosite (HBE) on FX. We determined the solution NMR structure and characterized the structural dynamics of the K1 and K2 domains of Ixolaris. Also, NMR data were used to construct a 3-dimensional structural model of the Ixolaris-FXa complex, revealing the noncanonical mechanism for FX binding, which occurs predominantly through the K2 domain. Taken together, our data provide new insight into the structural requirements for Ixolaris interaction with FX, which may contribute to our understanding of TF/VIIa inhibition.
Methods
Sample preparation
13C/15N and 15N-labeled Ixolaris samples were overexpressed in Escherichia coli strain Rosetta-gami B (DE3) as a TRX-His6 fusion protein and purified as described.27 U-[15N,2H]-labeled Ixolaris was overexpressed M9 medium in 2H2O containing 1 g/L 15NH4Cl and supplemented with Celtone Base Powder [U-15N,2H] 1 g/L (Cambridge Isotopes Laboratories). The final sample conditions for NMR spectroscopy were 0.1 to 0.2 mM Ixolaris, 20 mM sodium phosphate buffer (pH 6.0), 0.02% NaN3, 90/10% (vol/vol) H2O/D2O.
Human FX (58.8 kDa) was purchased from Enzyme research laboratories (catalog HFX 1010) as aliquots at concentration of 1.14 mg/mL in 20 mM Tris⋅HCl, 100 mM NaCl, 1 mM Benzamidine at pH 7.4. The FX was iteratively concentrated and buffer exchanged, using a Vivaspin Protein Concentrator centrifugal device (10 kDa molecular weight cutoff; GE). FX final sample concentration was 0.6 mM in 20 mM sodium phosphate buffer (pH 6.0), 0.02% NaN3.
Kunitz domains 1 and 2 expression and purification
Codon optimized DNA of isolated Kunitz domains were synthesized and cloned into the expression vector pET32a, as described.27 Both Kunitz domains K1 and K2 constructs were expressed in the E coli system according to the established protocol described for full-length Ixolaris. The purification of K1 and K2 domains included the combination of nickel affinity and reverse-phase chromatographies. Nonlabeled samples were dissolved in 20 mM sodium phosphate buffer at pH 6.0, 0.02% NaN3.
NMR assignments and dynamic measurements
All NMR experiments were recorded at 308 K, using Bruker DRX 600 equipped with a 1H,15N,13C TXI cryoprobe or Bruker Avance III 800 spectrometers. A set of 3D triple-resonance spectra was collected for sequential backbone and side-chain resonance assignments (De Paula et al28 ; BMRB: 27051). For obtaining nuclear Overhauser effect (NOE) connectivities, 3D [1H-13C-1H] and [1H-15N-1H] nuclear Overhauser spectroscopy (NOESY) spectra used for distance restrains were acquired with 120 ms mixing time. All spectra were processed using NMRpipe (Delaglio et al29 ) and analyzed with CARA 1.8.4.
For assessing the backbone dynamics, relaxation experiments were recorded on 0.18 mM 15N-labeled Ixolaris at a field of 600 MHz. 15N R1 and R2 relaxation rates were measured from spectra with different relaxation delays: 20, 50, 100 (duplicate), 200, 250, 500, 750, and 1000 ms for R1 and 48, 80 (duplicate), 112, 144, 176, 208, 240, 272, and 304 ms for R2. The errors in the peak intensities were calculated from the duplicate experiments. The 1H-15N heteronuclear NOEs were determined from the ratio of peak intensities with and without the saturation of the amide protons. Errors in the heteronuclear NOE values were calculated from the peak intensities and noise levels in the reference and saturated spectra. Relaxation data curve fitting was performed using CcpNmr analysis.30
Calculation of the NMR structure by the RASREC CS-Rosetta program
The CS-Rosetta/RASREC (resolution-adapted structural recombination) approach was used to generate the NMR structure of the Ixolaris. Briefly, assigned backbone chemical shifts (Cα, Cβ, N, and H) of the residues E13-S130 were used to predict backbone torsion angles (TALOS) and to generate a backbone fragment library representing the conformations of the protein.31 The N- and C-terminal residues were excluded, as they were identified as flexible according to the chemical shift-derived order parameter and heteronuclear NOE values. The Rosetta energy function was used to assemble and iteratively refine the structure.31,32 Unambiguous assigned long-range NOEs (i, i > 4) were used to bias the RASREC CS-Rosetta search. The RASREC protocol was used to generate a large pool of initial seed structures, which are then iteratively recombined, refined, and scored.33 The top 10 lowest-energy structures showing good Rosetta scores and agreement with the NOE and chemical shift data were selected as the final ensemble. The quality of structures was further checked by the Molprobity program.34 Structure calculations were performed at the University of California, Santa Cruz Baker cluster with the kind assistance of David Flores and Santrupti Nerli.
NMR titration and chemical shift mapping
For NMR binding experiments, FX (0.6 mM) was added progressively to a 15N Ixolaris sample (0.1 mM) in identical buffer conditions (20 mM sodium phosphate buffer at pH 6.0, 0.02% NaN3). For titration of FX into 15N Ixolaris, the following FX:Ixolaris molar ratios were used: 0.2:1, 0.5:1, and 1:1, and a series of 1H-15N HSQC spectra was collected. The pH at each step of the titration was kept constant. The chemical shift perturbations (CSPs) from the 2D 15N-1H HSQC NMR experiments were used to identify the FX binding site, using the following equation: ΔδNH = ([ΔH]2 + [Δ15N/10]2)1/2, where ΔH is the difference between free and bound 1H amide chemical shifts (ppm) and Δ15N is the difference between free and bound 15N chemical shifts (ppm). The change in peak intensity was determined by calculating the ratio of Ibound/Ifree for each assigned resonance and normalized to the dilution factor after FX addition. Chemical shift perturbations and peak intensities were calculated using CcpNmr Analysis.30
Molecular docking
Molecular docking of the Ixolaris to human FXa (Protein Data Bank [PDB] accession code 1hcg) was performed using HADDOCK 2.135-37 to generate an interaction model for this protein complex. The calculations were performed using NMR chemical shift mapping as ambiguous interaction restraints to define the residues potentially involved in binding. The active residues for Ixolaris were defined as the residues that had chemical shift changes above the mean plus 2 standard deviations (W15, G54, E55, K73, Y94, D95, N97, E119), and the passive residues the ones having resonances intensity significantly attenuated (V16, S17, C18, L19, Y36, G41, H57, F58, N65, E66, S67, C68, W92, T96, F104, T105, N115, K123, E124, T125, K127) in the NMR titrations between Ixolaris and FX (Figure 3).
Active residues for FXa included R93, K96, R125, R165, K169, K236, and R240, and were defined from previous mutagenesis experiments.21 Passive residues were automatically defined by HADDOCK. Complex structures were sorted according to the intermolecular interaction energy (the sum of intermolecular van der Waals and electrostatic energies and restraint energies). The calculated models were clustered using a 7.5-Å interface root mean square deviation cutoff. HADDOCK clustered 146 of 200 calculated structures into 12 clusters. The top HADDOCK cluster with the lowest energy structure within the highest-score NMR-restrained were considered to be the most reliable Ixolaris-FXa complex.
Results
Structure determination of Ixolaris obtained by NMR and RASREC CS-Rosetta
Ixolaris is a monomeric protein in solution, and exhibits a well-resolved 1H-15N heteronuclear single quantum coherence (HSQC) NMR spectrum that is typically observed in compact folded proteins (supplemental Figure 1, available on the Blood Web site). The 1H, 13C, and 15N resonance assignments and secondary structure information of Ixolaris based on 13C chemical shifts were determined previously (De Paula et al28 ). Although they were stable and give good spectra, Ixolaris structure calculations using NMR data were a challenge and required a different strategy that applied NOE and chemical shift information.
Our first attempt was to calculate the structure with conventional methods, using dihedral angles, disulfide bonds, and all NOEs from the 3-dimensional [1H-13C-1H] and [1H-15N-1H] NOESY spectra. We used semiautomated NOE cross-peak assignments, using the UNIO’10 module38-40 and the simulated annealing protocol with torsion angle molecular dynamics with CYANA version 2.1.41 However, we obtained nonconvergent structures that showed very large backbone root mean square deviation (RMSD) values resulting from the small number of unambiguous NOEs in the 3-dimensional [1H-13C-1H] NOESY spectra. Toward obtaining a more converged structural model of Ixolaris from our sparse NOE data (38 unambiguously assigned long-range NOEs), we turned to the RASREC CS-Rosetta. In the Rosetta methodology, a limited set of NMR data are sufficient to accurately determine solution structures of proteins up to 40 kDa.32,33,42,43 We calculated 100 Rosetta structures for the core region of Ixolaris (residues 13-130) and selected the 10 lowest-energy structures for further analysis (Figure 1). The family of the 10 lowest-energy structures of Ixolaris is well defined, with an average backbone pairwise RMSD of 1.39 Å (Figure 1A) and corresponding RMSDs for the K1 and K2 domains in isolation of 0.50 and 0.40 Å, respectively (supplemental Figure 3). The final full-atom models have good packing and structural statistics, as assessed using MOLPROBITY, which satisfy all experimental NOE restraints (Table 1).
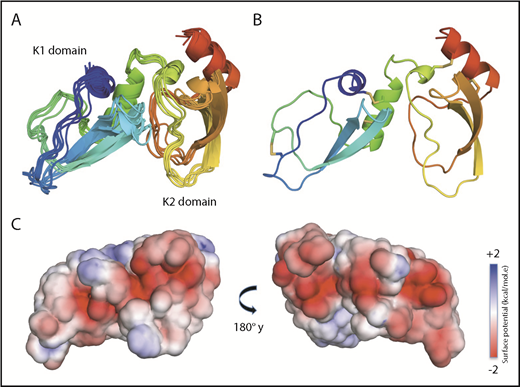
NMR solution structure of Ixolaris. (A) Ribbon diagrams of the 10 lowest-energy models of the Ixolaris calculated by the CS-Rosetta/RASREC program with chemical shifts and long-range NOEs. The ensemble is converged to an average backbone RMSD of 1.39 Å over the core residues. (B) The lowest-energy structure of Ixolaris. The 5 disulfide bonds are shown as sticks. (C) Solvent-accessible surface representation with electrostatic potential −2 kcal/(mol·e) in red and +2 kcal/(mol·e) in blue for Ixolaris as calculated in CHARMM-GUI PBEQ-Solver44 (PDB accession code 6NAN).
NMR solution structure of Ixolaris. (A) Ribbon diagrams of the 10 lowest-energy models of the Ixolaris calculated by the CS-Rosetta/RASREC program with chemical shifts and long-range NOEs. The ensemble is converged to an average backbone RMSD of 1.39 Å over the core residues. (B) The lowest-energy structure of Ixolaris. The 5 disulfide bonds are shown as sticks. (C) Solvent-accessible surface representation with electrostatic potential −2 kcal/(mol·e) in red and +2 kcal/(mol·e) in blue for Ixolaris as calculated in CHARMM-GUI PBEQ-Solver44 (PDB accession code 6NAN).
Statistics for 10 low-energy Ixolaris models
| Number of NMR distance restraints | 38 |
| Restraints eliminated because of distance violations* | |
| Between 1 and 2 Å/structure† | 0.4 ± 0.4 |
| Above 5.0 Å/structure | 0.0 ± 0.0 |
| Number of disulfide restraints‡ | 5 |
| Ramachandran statistics¶ | |
| % of residues in favored regions | 91.9 |
| % of residues in allowed regions | 99.9 |
| Outliers | 0 |
| Average RMSD to mean structure (Å)§ | |
| Backbone atoms | 1.39 ± 0.58 |
| Heavy atoms | 1.53 ± 0.63 |
| Number of NMR distance restraints | 38 |
| Restraints eliminated because of distance violations* | |
| Between 1 and 2 Å/structure† | 0.4 ± 0.4 |
| Above 5.0 Å/structure | 0.0 ± 0.0 |
| Number of disulfide restraints‡ | 5 |
| Ramachandran statistics¶ | |
| % of residues in favored regions | 91.9 |
| % of residues in allowed regions | 99.9 |
| Outliers | 0 |
| Average RMSD to mean structure (Å)§ | |
| Backbone atoms | 1.39 ± 0.58 |
| Heavy atoms | 1.53 ± 0.63 |
Violations were calculated using 5 Å universal upper bound distance for structurally degenerate and unambiguous NOE restraints
One NOE has violated in 4 structures.
Disulfide restraints were used between the pairs of residues: 18-68, 26-51, 43-64, 76-126, and 101-122.
Ramachandran statistics were calculated using MOLPROBITY over the low-energy models.
Computed over 10 low-energy structures for core residues 2-7, 20-25, 29-35, 49-58, 76-83, 87-94, 107-115.
As previously predicted from sequence homology analysis, the structure of Ixolaris confirms the presence of 2 consecutive Kunitz domains. Each of the 2 domains of Ixolaris displays the characteristic fold of the Kunitz family of protease inhibitors, which is composed of a central antiparallel β sheet (residues 32-36, 43-45, 91-95, and 104-107) with 1 short α-helix at the C-terminal end (residues 61-68 and 119-125). In addition, a short α-helix (residues 13-18) is formed at the N-terminal of the K1 domain. These elements form the topological scaffold that is stabilized by the 5 canonical disulfide bonds involving Cys18-Cys68, Cys26-Cys51, Cys43-Cys64, Cys76-Cys126, and Cys101-Cys122. Analysis of the 13Cα and 13Cβ chemical shifts of the 10 cysteines confirms their oxidized state. Additional stabilization derives from a remarkably hydrophobic cluster formation in the K2 domain, formed by WYY. A triple aromatic residue cluster is highly conserved in Kunitz inhibitors (supplemental Figure 3). Ixolaris is an acidic protein (pI ∼4.6), and the surface electrostatic potential indicates that negative charges are densely distributed in the K1 and K2 domains (Figure 1C).
As seen in other Kunitz domain proteins, only ∼53% of the structure is engaged in secondary structure elements, which are connected by long loops (residues 46-60, 69-90, 108-118; Figure 1). In addition, the loop region spanning residues 71 to 75 is the peptide linker between the first and second Kunitz domains of Ixolaris. This linker contains 3 prolines and 1 residue (K73) mobile on the picosecond to nanosecond timescale, as evidenced by the reduced 15N-1H NOE and R2 values (Figure 2A). The most similar structure found by structural alignment software (PDB accession code efold) belongs to the Kunitz inhibitors, including Bikunin (PDB accession code 1BIK), APPI (Amyloid Precursor Protein Inhibitor; PDB accession code 1CA0), BPTI (Bovine Pancreatic Trypsin Inhibitor; PDB accession code 1MTN), and TFPI (Tissue Factor Pathway Inhibitor; PDB accession code 4BQD). All these structures displayed RMSD between 1.65 and 3.55 Å, with sequence identity ranging between 20% and 27%.

Backbone dynamics of Ixolaris. (A) Backbone 15N relaxation parameters and heteronuclear 1H- 15N NOEs for Ixolaris at 35°C at a 1H field of 600 MHz. All data were recorded using a 0.15 mM 15N,13C labeled Ixolaris in the NMR buffer (20 mM phosphate buffer at pH 6.0) and are consistent with a predominantly monomeric form with a rotational correlation time of 6.6 ns/rad. The absences of data indicate that the values could not be accurately measured because of overlaps or are prolines, or residues not assigned. Gray boxes highlight dynamic regions of interest. R2 ratios values 1 standard deviation above the trimmed mean, are indicative of conformational exchange. Residues with heteronuclear NOE ratios below 0.6 suggest that they possess fast internal motion. The secondary structure diagram based on the Talos+ score is shown at the top as a guide. (B) Residues with NMR resonances exhibiting dynamics are indicated on the structure of Ixolaris as orange (slow exchange) and green (fast internal motions) spheres.
Backbone dynamics of Ixolaris. (A) Backbone 15N relaxation parameters and heteronuclear 1H- 15N NOEs for Ixolaris at 35°C at a 1H field of 600 MHz. All data were recorded using a 0.15 mM 15N,13C labeled Ixolaris in the NMR buffer (20 mM phosphate buffer at pH 6.0) and are consistent with a predominantly monomeric form with a rotational correlation time of 6.6 ns/rad. The absences of data indicate that the values could not be accurately measured because of overlaps or are prolines, or residues not assigned. Gray boxes highlight dynamic regions of interest. R2 ratios values 1 standard deviation above the trimmed mean, are indicative of conformational exchange. Residues with heteronuclear NOE ratios below 0.6 suggest that they possess fast internal motion. The secondary structure diagram based on the Talos+ score is shown at the top as a guide. (B) Residues with NMR resonances exhibiting dynamics are indicated on the structure of Ixolaris as orange (slow exchange) and green (fast internal motions) spheres.
K2 domain is particularly dynamic
To gain insight into the behavior of Ixolaris in solution, we measured 15N relaxation parameters to probe dynamics on the fast (pico- to nanoseconds) and intermediate (micro- to milliseconds) timescale. We recorded R1, R2, and heteronuclear 15N-1H NOE experiments at 600 MHz and 308 K (Figure 2). On the basis of the NMR relaxation data, Ixolaris backbone is well structured (with the exception of the N- and C-terminal residues), with an average tumbling rate of 6.6 ns, which is typical for the size of Ixolaris (∼16 kDa). The heteronuclear 15N-{1H} NOE, which report specifically on backbone fluctuations on a pico- to nanosecond (ps-ns) timescale, show several residues of the K2 domain with significantly lower NOE values, indicating a higher degree of flexibility when compared with the K1 domain (Figure 2A).
In addition, our data indicate the presence of residues with motions in the intermediate timescale (higher R2 values), indicative of conformational exchange in the K1; however, most notably, in the K2 domain. This suggests an evident distinction between the K1 and K2 dynamics. The R2 values for several residues in the K2 domain (G83, Y94, D95, T96, T105, Y106, E119) were higher than the average, typical of internal motion on a micro- to millisecond timescale. It is important to mention that a few amide groups were not assigned (R3, F12, E45, Y82, V84, R86, I89, C101, T110, G111, N112, K113, and N114), which indicates local motion in the intermediate exchange regime. The majority of those residues are located at the K2 domain.
A summary of the Ixolaris dynamics is shown in Figure 2B, from which it is evident that the K2 domain is particularly dynamic, whereas the K1 domain keeps as a rigid platform supporting the conformational dynamic of the K2 domain.
NMR reveals FX-binding effects on the Ixolaris structure
The backbone assignment of free Ixolaris allowed us to probe conformational changes on binding to FX. The complex between Ixolaris and FX is 74.7 kDa, and is consequently difficult to be characterized by standard NMR methods. In agreement with previous studies,21,22 Ixolaris formed a tight complex with FX under the NMR conditions. Detailed inspection of the [15N, 1H] HSQC spectra of Ixolaris titrated with unlabeled FX at different ratios showed very little perturbation of chemical shifts, indicating that the binding process is intermediate to slow on the NMR timescale (Figure 3A). In addition, most amide resonances of Ixolaris exhibited an intensity reduction of ∼60% (low Ibound/Ifree values), when the complex formation reached 50% from the stoichiometric titration (Figure 3C). This indicates that the line broadening is caused not only by the large complex formation but also by intermediate exchange from the interaction. Further addition (1:1 molar ratio) resulted in the significant line broadening, except for the flexible N- and C-terminal regions (supplemental Figure 4). As such, we evaluated the changes in peak positions and intensities in the presence of FX at a molar ratio of 1:0.5 (Ixolaris:FX). Analysis revealed 2 types of effects on the Ixolaris amide resonances: chemical shift perturbations (Figure 3B, pink bars), indicating a change in the local chemical environment, and conformational exchange-induced line broadening in the bound state (Figure 3C).

NMR characterization of Ixolaris-FX complex using 15N probes. (A) Overlays of representative regions from 2-dimensional 15N,1H HSQC spectra recorded in the free (magenta) and 0.5:1 FX-bound (black) state of the Ixolaris. (B) Histogram of CSPs and (C) peak intensity ratio (Ibound/Ifree) as a function of Ixolaris residue number. The dashed line indicates the magnitude of the CSPs corresponding to the 2 standard deviation greater than the mean. (D) Mapping of amides probes on the NMR structure of Ixolaris with K1 domain colored wheat and K2 domain colored light blue. Two types of observed changes are highlighted with different colors: residues with significant chemical shift changes are in magenta spheres (>2 standard deviation from the average), whereas residues with significantly attenuated intensity resonances (>1 standard deviation from the average) in the bound form are shown in cyan spheres.
NMR characterization of Ixolaris-FX complex using 15N probes. (A) Overlays of representative regions from 2-dimensional 15N,1H HSQC spectra recorded in the free (magenta) and 0.5:1 FX-bound (black) state of the Ixolaris. (B) Histogram of CSPs and (C) peak intensity ratio (Ibound/Ifree) as a function of Ixolaris residue number. The dashed line indicates the magnitude of the CSPs corresponding to the 2 standard deviation greater than the mean. (D) Mapping of amides probes on the NMR structure of Ixolaris with K1 domain colored wheat and K2 domain colored light blue. Two types of observed changes are highlighted with different colors: residues with significant chemical shift changes are in magenta spheres (>2 standard deviation from the average), whereas residues with significantly attenuated intensity resonances (>1 standard deviation from the average) in the bound form are shown in cyan spheres.
Mapping the FX-induced chemical shift perturbations onto the Ixolaris structure revealed a continuous and extensive interaction surface (Figure 3D). Relevant residues of K2 domain appear to contribute to the binding of FX (13 residues); however, only residues W92, Y94, F104, N115, and K123 are conserved in TFPI K2 domain (sequence alignment shown in supplemental Figure 3). In addition, the peak intensities of residues located in the H3-helix of K2 domain were also significantly attenuated in the bound form. Interestingly, the NMR data revealed effects on the K1 domain as well. These changes are highly localized, and cluster near the H2-helix (N65, E66, S67, C68, K73) at the interface with the K2 domain (Figure 3D). Notably, residues W15, V16, S17, C18, L19, G54, E55, H57, and F58, located at or near the H1-helix, also showed 2 standard deviation above average chemical shift perturbations and attenuated peak intensities, indicating that FX-binding-induced conformational changes are not restricted to the K2 domain. The perturbed residues on Ixolaris indicate that polar and hydrophobic interactions contribute for FX binding.
Remarkably, we found that the Ixolaris residues involved in FX association, specifically from the K2 domain, correlate with regions of increased dynamics (Figure 2), indicating a direct role for dynamics in driving the recognition of the inhibitors for the blood coagulation factor.
Structural model for the Ixolaris/factor Xa complex
To derive a structural model for the Ixolaris/FXa complex binding site, we integrated the information that was obtained in the current work (Figure 4) with analysis obtained in a previous study showing that residues R93, K96, R125, R165, K169, K236, and R240 from FX(a) are critical for Ixolaris/FX(a) binding.21 To this end, the lowest energy conformation of Ixolaris and the crystal structure of human FXa were used as starting structures for the docking process. Ixolaris was docked to the solvent-exposed, hydrophilic FXa HBE,20 using the software HADDOCK.35-37 The lowest-energy structure obtained from the restrained docking of the Ixolaris-FXa complex (Figure 4A) was consistent with the NMR data, and the interface agrees with our experimental data. The Ixolaris-FXa interface buries a total of 1707 Å2 of accessible surface area, with both domains contributing to a polar interaction to the hydrophilic pocket of FXa (Figure 4A; supplemental Figure 4). Ixolaris and FXa display pronounced electrostatic complementarity spanning the entire HBE, with Ixolaris presenting a negatively charged surface patch associating with the positively charged HBE of FXa (Figure 4B). This suggests that long-range electrostatic interactions may play an important role in attracting Ixolaris to FXa to establish the binary complex.

Model structure of the Ixolaris-FXa complex. NMR data-driven HADDOCK model structure of Ixolaris bound to FXa. (A) Model with lowest HADDOCK score with FXa shown as green ribbon and Ixolaris as light blue and wheat ribbon. The model is based on the FXa structure (PDB accession code 1hcg) and the amino acids identified to interact with Ixolaris were used as active residues. The Ixolaris residues identified to interact with FXa using NMR CSP and peak intensity are shown as in Figure 3D. The orientation of membrane-associated FXa by the EGF and Gla domains is shown. The FXa catalytic triad (H57, D102, and S195) is shown as orange sticks. BSA, buried surface area. (B) Side view cartoon representations of the Ixolaris-FXa complex with the electrostatic potential at the solvent accessible surfaces shown for FXa (top) and Ixolaris (bottom). (C) Differential scanning fluorimetry of FX free state and FX bound state incubated with Ixolaris full-length, K1, and K2 constructs (molar ratio 1:1). Analysis of these melting curves with a first derivative method delivered the corresponding Tm values that were used to calculate the shifts (ΔTm).
Model structure of the Ixolaris-FXa complex. NMR data-driven HADDOCK model structure of Ixolaris bound to FXa. (A) Model with lowest HADDOCK score with FXa shown as green ribbon and Ixolaris as light blue and wheat ribbon. The model is based on the FXa structure (PDB accession code 1hcg) and the amino acids identified to interact with Ixolaris were used as active residues. The Ixolaris residues identified to interact with FXa using NMR CSP and peak intensity are shown as in Figure 3D. The orientation of membrane-associated FXa by the EGF and Gla domains is shown. The FXa catalytic triad (H57, D102, and S195) is shown as orange sticks. BSA, buried surface area. (B) Side view cartoon representations of the Ixolaris-FXa complex with the electrostatic potential at the solvent accessible surfaces shown for FXa (top) and Ixolaris (bottom). (C) Differential scanning fluorimetry of FX free state and FX bound state incubated with Ixolaris full-length, K1, and K2 constructs (molar ratio 1:1). Analysis of these melting curves with a first derivative method delivered the corresponding Tm values that were used to calculate the shifts (ΔTm).
The observation that both K1 and K2 domains of the Ixolaris interact with human FX, we sought to characterize the relative contributions of the isolated domains toward the overall interaction. Using thermal stability shift assay, we measured the FX thermal stability on complex formation (Figure 4C; supplemental Figure 6). Ixolaris binding to FX results in a significant thermal stabilization of the protein: ΔTm shift of 10°C. Interestingly, no stabilization was observed for both K1 and K2 single domains. In another sample, K1 and K2 single domains added simultaneously (up to 3:1 molar ratio) into FX also did not affect the Tm (supplemental Figure 6). Together, these results show that FX exhibits greater thermal stability on Ixolaris binding relative to association with single domains, and both K1 and K2 domains are necessary and must reside in the same polypeptide for binding to FX.
Structure of the Ixolaris-FXa complex reveals an ‘‘allosteric switch’’
Additional insights into the possible noncanonical mechanism of Ixolaris are provided by the HADDOCK structure analysis. Close inspection of the specific interactions between Ixolaris and FXa involved salt bridges between residues E22, E55, K73, D95, and T96 of Ixolaris and residues R93, R240, E129, N166, and K169 of FXa, respectively (Figure 5A).
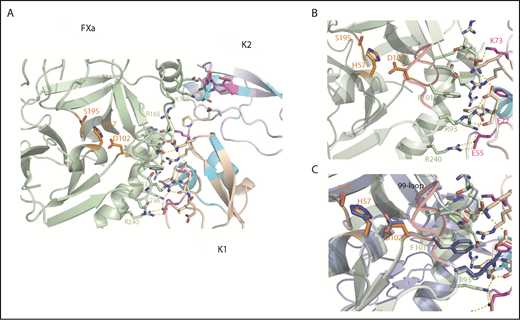
Structural snapshots of the Ixolaris-FXa complex and insights into the allosteric mechanism. (A) Top view and expansion of Ixolaris bound to FXa. Ixolaris-FXa contacts within 3Å in the HADDOCK model structure, focusing on the HBE. Residues in FXa that are in direct contact in the structure are shown in green sticks and labeled. Ixolaris residues that are in direct contact in the structure and affected in NMR experiments are shown in magenta sticks. Hydrogen bonds and salt bridges formed with the arginines and lysines in the HBE (obtained by HADDOCK output) are shown as yellow dashed lines. (B) Detail of FXa-K1 domain interface. Critical residues involved in the interface interactions are highlighted. (C) Superposition of Ixolaris/FXa (light green) binary complex with FXa apo state (blue) showing the conformation transition of the 99-loop (red), H57, D102, and R93 side chains on Ixolaris binding.
Structural snapshots of the Ixolaris-FXa complex and insights into the allosteric mechanism. (A) Top view and expansion of Ixolaris bound to FXa. Ixolaris-FXa contacts within 3Å in the HADDOCK model structure, focusing on the HBE. Residues in FXa that are in direct contact in the structure are shown in green sticks and labeled. Ixolaris residues that are in direct contact in the structure and affected in NMR experiments are shown in magenta sticks. Hydrogen bonds and salt bridges formed with the arginines and lysines in the HBE (obtained by HADDOCK output) are shown as yellow dashed lines. (B) Detail of FXa-K1 domain interface. Critical residues involved in the interface interactions are highlighted. (C) Superposition of Ixolaris/FXa (light green) binary complex with FXa apo state (blue) showing the conformation transition of the 99-loop (red), H57, D102, and R93 side chains on Ixolaris binding.
Most notably, E55 in the K1 domain makes a salt bridge with K236 and R240 of FXa (Figure 5B). Furthermore, structural superposition of FXa in its apo and Ixolaris-bound state shows that the R93 and residues H57 and D102 of the catalytic triad adopt different conformations (Figure 5C). The closest atom of Ixolaris is more than 15 Å from the active site Ser195 residue, indicating an allosteric switch in the catalytic site. No other significant changes in the FXa structure were observed, with the rest of the FXa main chain being very similar (RMSD of 0.46 Å for 210 aligned Cα atoms). Thus, the data support our hypothesis that the mechanism of allosteric modulation by Ixolaris is primarily driven by a change of the FXa 99-loop conformation (Figure 5C). The 99-loop modulates the protease activity by both hindering the substrate binding and keeping the proper arrangement of the catalytic triad. In addition, loop flexibility was observed in 2 trypsin-like serine proteases.45,46
Discussion
In this study, we have employed solution NMR to directly evaluate the plasticity and conformational dynamics to propose a mechanistic blueprint for Ixolaris binding to scaffold FX, a step critical for inhibition of the TF/FVIIa complex. The basis of our mechanistic approach is the high-affinity Ixolaris-FXa encounter complex driven by long-range electrostatic attraction of Ixolaris to the FXa HBE, revealing a noncanonical mechanism, mainly conducted by the K2 domain. We therefore hypothesize that the negatively charged and dynamic region on K2 domain is the major binding site for the positively charged FXa HBE. In addition to the major contribution of the K2 domain, a secondary interface formed by the K1 domain is necessary for the FXa binding. More specifically, E22 of the K1 domain makes a salt bridge with R93 of the FXa, possibly revealing a conformational change at the 99-loop that could allosterically modulate the FXa catalytic site (Figure 5B). Notably, a previous study has demonstrated that R93 is critical for the recognition of the HBE by Ixolaris. In addition, the interaction of Ixolaris with R93-mutated FXa is by far the most severely impaired among all mutants tested.21 The presence of a secondary site in the complex possibly creates a loss of entropy in the binding that supports the interaction of the K2 domain and contributes to the stabilization of the complex. In addition, Ixolaris-FXa model highpoints a second key concerted structural event, in which the K1 domain could simultaneously interact with both FXa exosite and FVIIa/TF active site. Accordingly, prior in vitro studies have shown that Ixolaris inhibition of FVIIa catalytic activity is markedly reduced in T99 and Q143-mutated FVIIa.23
To determine unique features of the Ixolaris in relation to other Kunitz domains proteins, we compared the structures of the K2 domain of Ixolaris, TFPI (1TFX), and Bikunin (1BIK) (supplemental Figure 7). The 3 domains showed high similarity with a backbone RMSD of 0.96 Å (Bikunin) and 1.2 Å (TFPI). The structural signature of the canonical Kunitz domains was observed in Ixolaris, despite the fact that 1 bridge is missing (supplemental Figure 3). The loops D81-I89 and I109-N115 without the canonical disulfide bridge differ only slightly from the arrangement in TFPI. In addition, we observed that the majority (56%) of the amide backbone chemical shifts for these loops were not assigned, which indicates local motions in the intermediate exchange regime.
These structural features in Ixolaris may affect the conformational mobility of its K2 domain, which merit further analysis in extended structural studies. To determine whether this observation is part of a unique mechanism of Ixolaris, it would be interesting to confirm the existence of similar conformational mobility in the related Kunitz structures by measuring their dynamics as well.
Ixolaris shares a similar anticoagulant mechanism with hematophagous nematode NAPc2 with respect to scaffold requirements for inhibition of TF/FVIIa complex. However, NAPc2 uses a different exosite in FXa to inhibit TF-FVIIa.47 Structural analyses of the Ixolaris-bound FXa, NAPc2-bound FXa, and Apo FXa structures suggest that residues H57 and D102 of the catalytic triad exhibit conformational changes when bound to Ixolaris, whereas minor changes were observed for NAPc2-bound FXa (supplemental Figure 8). Given that NAPc2 blocks the active site of FVIIa, while locking in FXa in a signaling active conformation on the ternary TF-FVIIa-FXa complex, it seems reasonable to speculate that the small changes in the catalytic triad could render the FXa unable to be an active protease, perhaps explaining why Ixolaris blocks PAR2 cleavage and NAPc2 does not.23
In contrast to a previous suggestion,47 our NMR data showed that the C-terminal of Ixolaris is not involved in HBE binding. Slightly different results were obtained with NAP5 from Ancylostoma caninum. This inhibitor binds to both the catalytic site and the HBE of FXa.48 Ixolaris is also distinct from the Kazal-type inhibitor Rhodniin, which employs its N- and C-terminus domains to interact simultaneously with the thrombin catalytic site and the anion-binding exosite, respectively.49
Although Ixolaris shows a canonical Kunitz 3-dimensional structure, it exhibits a noncanonical mechanism of inhibition characterized by interaction outside the active site of FX, which operates as an efficient scaffold for formation of a tight quaternary inhibited FVIIa/TF/Ixolaris/FX(a) complex.19 This step is critical to understand how a binary Ixolaris/FX complex blocks FVIIa/TF. Accordingly, this study contributes to the development of specific inhibitors targeting nonconserved binding sites outside the active center. These domains are critical in the specificity and productive interaction of enzymes, cofactors, and zymogens, and are therefore potential targets for the inhibition of coagulation factors.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The structure of Ixolaris has been deposited in the Protein Data Bank under accession code 6NAN. The NMR assignments for Ixolaris have been deposited in the Biological Magnetic Resonance Data Bank under accession code 27051.
For original data, please contact valente.anap@gmail.com.
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by grants from Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro and Conselho Nacional de Desenvolvimento Científico e Tecnológico. The authors acknowledge the University of California, Santa Cruz Baker cluster for providing access to the computer resources. The authors also thank the facilities of National Center of Nuclear Magnetic Resonance (Federal University of Rio de Janeiro, Rio de Janeiro) for the NMR time.
Authorship
Contribution: V.S.D.P., R.Q.M., and A.P.V. designed the research and wrote the manuscript. V.S.D.P. and A.P.V. performed and analyzed the NMR experiments and NMR data. V.S.D.P. generated constructs, prepared and purified isotopically labeled proteins; assigned NOEs; performed structure calculations with Cyana, RASREC CS-Rosetta, and HADDOCK; performed differential scanning fluorimetry; and analyzed and interpreted all data. V.S.D.P., I.M.B.F., R.Q.M., and A.P.V. provided reagents and analytical tools. N.G.S. and F.C.L.A. contributed with CS-Rosetta calculations. All authors discussed the results and contributed to writing the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ana Paula Valente, Instituto de Bioquímica Médica, Centro Nacional de Ressonância Magnética Nuclear Jiri Jonas, Universidade Federal do Rio de Janeiro, Brazil; e-mail: valente.anap@gmail.com; and Viviane Silva de Paula, Department of Chemistry and Biochemistry, University of California, Santa Cruz, CA 95064; e-mail: vsilvade@ucsc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal