

Transcription factor RUNX1 plays important roles in hematopoiesis and leukemogenesis. As a master regulator for hematopoiesis, RUNX1function is tightly controlled through posttranslational modifications, including ubiquitination. We and others have identified multiple E3 ubiquitin ligases that promote RUNX1 ubiquitination, such as STUB1, RNF38 and APC complex. However, mechanisms for ubiquitination-mediated regulation of RUNX1 function, especially the proteasome-independent roles of ubiquitination, have not been fully understood.
We previously established a high-throughput AlphaScreen assayto measure the interaction between RUNX1 and E3 ubiquitin ligases.In this assay, biotinylated full-length RUNX1 was captured by streptavidin-coated donor beads and FLAG-tagged E3 ligases were bound by protein A-conjugated acceptor beads. The binding of RUNX1 to each E3 ligase brings the donor and acceptor beads in close proximity, enabling energy transfer between them, resulting in a chemiluminescence signal. This cell-free AlphaScreen assay allows for the efficient detection of RUNX1-interacting E3 ligases with high sensitivity, and have revealed several E3 ligases, including DTX2, that interact with RUNX1.
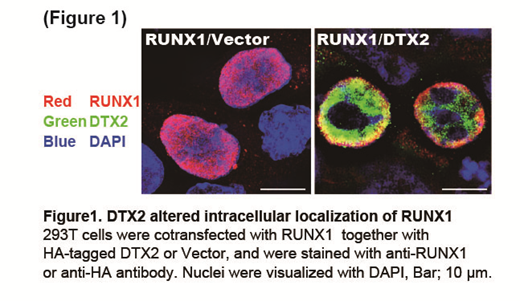
DTX2 is a mammalian homolog of Drosophila deltex and functions as a RING-finger E3 ubiquitin ligase. Interestingly, a RUNX1 fusion gene connected to reversed DTX2 was identified in acute myeloid leukemia with t(7;21). Given that the reversed sequence of DTX2could inhibit activities of wild-type DTX2, the presence of RUNX1-DTX2 (reversed-sequence) chimera in AML indicates the functional association between RUNX1 and DTX2 in hematopoietic cells. To test this possibility, we first examined whether DTX2 physically interacts with RUNX1 to promote its ubiquitination. Immunoprecipitation assays revealed that DTX2 in fact interacted with RUNX1 in 293T cells. We also found that DTX2, but not a DTX2 mutant lacking Ring finger domain (DTX2-DR), induced ubiquitination of RUNX1 in both nucleus and cytoplasm. However, DTX2-induced RUNX1 ubiquitination did not result in degradation of RUNX1 protein, suggesting that DTX2 modifies RUNX1 function mainly through a proteasome-independent mechanism. We therefore examined if DTX2 alters subcellular localization of RUNX1 using immunofluorescence analysis. Strikingly, ectopic expression of DTX2 altered RUNX1 distribution from nuclear towards cytoplasm in 293T cells (Figure 1). Furthermore, we observed similar cytoplasmic localization of endogenous RUNX1 in DTX2-transduced K562 cells. We then assessed whether DTX2 is involved in RUNX1-mediated transcriptional regulation using the MCSFR luciferase reporter, and found that RUNX1-induced reporter activation was inhibited by ectopic expression of DTX2. Taken together, these data suggest that DTX2 promotes nuclear export of RUNX1, thereby inhibits its transcriptional activity.
Next, we assessed the effect of DTX2 on RUNX1 function in primary human cord blood cells. In agreement with previous reports, RUNX1 overexpression promoted myeloid differentiation and downregulated a stem cell marker CD34 in cord blood cells. This RUNX1-induced CD34 downregulation was attenuated by DTX2 coexpression, suggesting the inhibitory effect of DTX2 on RUNX1 activity. We then examined the effect of DTX2 expression in several leukemia cell lines: Jurkat, TF-1, Kasumi-1, SKNO-1 and K562 cells. Previous studies have shown that Jurkat, TF-1, Kasumi-1 and SKNO-1 cells are dependent on RUNX1 activity for their growth. In contrast, K562 cells were shown to be relatively resistant to RUNX1 depletion. Consistent with our earlier results, expression of DTX2 promoted cytoplasmic localization of RUNX1 in all these leukemia cell lines. Functionally, DTX2 overexpression inhibited the growth of RUNX1-dependent leukemia cell lines (Jurkat, TF-1, Kasumi-1, and SKNO-1) by inducing apoptosis and cell-cycle arrest, while it showed only modest effect in K562 cells. Thus, these results indicate again that DTX2 inhibit RUNX1 function by promoting its cytoplasmic localization in hematopoietic and leukemia cells.
In summary, we identified DTX2 as an E3 ligase to promote RUNX1 ubiquitination. DTX2 does not induce RUNX1 degradation, but instead promotes nuclear export of RUNX1 to inhibit its function. Activation of DTX2 could be a therapeutic approach to treat RUNX1-dependent leukemias.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal