

FLT3-ITD mutations are seen in 20-30% of adult and 15% of pediatric acute myeloid leukemia (AML) patients and are associated with poor prognosis with higher rates of relapse and therapy resistance. FLT3 tyrosine kinase inhibitors (TKIs) have shown some success in treatment of FLT3-ITD AML, but responses are often not complete or sustained. Multiple potential mechanisms of drug resistance could be active in patients with FLT3-ITD AML including mutations in the FLT3 kinase, exogenous signaling from the bone marrow microenvironment, and altered epigenetic regulation. Mutations involving epigenetic modifiers such as DNMT3A, IDH1/2, TET2, ASXL1 and MLL1 are often co-expressed with the FLT3-ITD mutation. In addition, over-expression of EZH2, DOT1L and protein arginine methyl-transferase (PRMT) have also been reported. However, the role of different epigenetic mechanisms in regulating growth of FLT3-ITD AML cells and to modulate ability of TKIs to target FLT3-ITD AML cells has not been systematically studied. The objective of our study was to identify epigenetic targets in FLT3-ITD AML by screening a panel of epigenetic modifiers for their ability to inhibit growth of FLT3-ITD AML cells, and to enhance growth inhibition in combination with a FLT3 TKI.
We used a carefully curated and well validated collection of epigenetic inhibitors (Structural Genomics consortium, Epigenetics Probes Collection, SGC Toronto; www.thesgc.org) that included 38 probes that target a spectrum of proteins involved in epigenetic regulation. We exposed MOLM-13 and MV4-11 human FLT3-ITD positive AML cells to each inhibitor by itself and in combination with the TKI Quizartinib/AC220 (500pM, IC20). Cell growth was measured using the CellTiter-Glo® Luminescent Cell Viability Assay. The FLT3-ITD negative AML cell line OCI-AML3 (FLT3-WT) was studied as a control. The combined results of three experiments carried out using triplicate wells in each experiment were analyzed. Apoptosis was measured by flow cytometry using Annexin-V/FITC labeling and data analyzed by FlowJo V10. Statistical analysis was performed using GraphPad Prism 8.1.1 software.
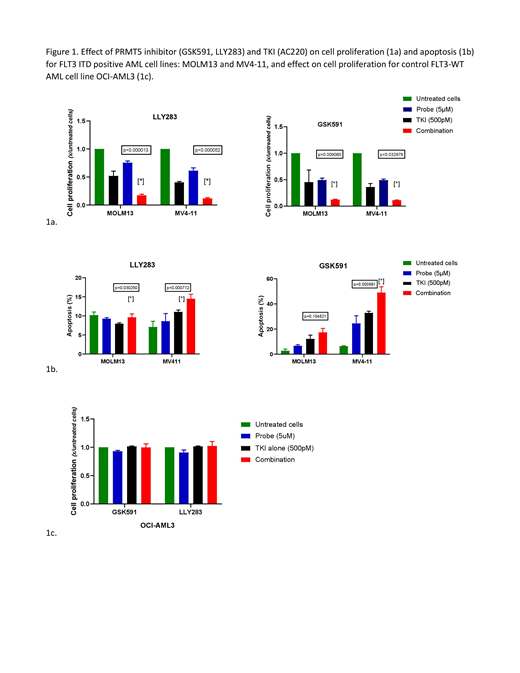
Of a total of 38 epigenetic inhibitors, less than a third resulted in reduction in cell proliferation after 72 hours of treatment (greater than 10% decrease, 13 for MOLM13, 9 for MV4-11 cell line). When used in combination with TKI (AC220), several epigenetic inhibitors resulted in significantly increased inhibition of proliferation of FLT3-ITD positive AML cells compared to either agent alone (21 for MOLM13, 10 for MV4-11 cell line) (p<0.05). These included several Bromodomain inhibitors (L-moses, SGC-CBP30, OF1), EZH2/H1 inhibitors (GSK343, UNC1999) and PRMT1 and PRMT5 inhibitors (MSO23, GSK591, LLY283). The combinatorial effect was not seen in the control FLT3-WT OCI-AML3 cells. Given the selective effect of the combination of PRMT5 and TKI on FLT3-ITD positive AML cells in our screen, and reports of over-expression of PRMT5 in hematologic malignancies and its described direct influence on FLT3 gene transcription, we further studied the effect of PRMT5 inhibitors in combination with TKI on FLT3-ITD positive AML cells. The effectiveness of the combination of PRMT5 inhibitor and TKI in MOLM13 and MV4-11 cells but not OCI-AML3 cells was confirmed by fixed-ratio dose-response analysis. We further show that the combination of PRMT5 inhibitors (GSK591, LLY283 5μM) with AC220 (500pM) resulted in increased inhibition of cell proliferation and increased induction of apoptosis compared to TKI alone, p<0.05 (Figure 1a-c).
In conclusion, our studies using an unbiased approach have identified PRMT5 inhibition as a novel and selective approach to enhance targeting of FLT3-ITD AML cells in combination with FLT3 TKI. These results support our ongoing studies to evaluate the cellular and molecular mechanisms underlying these combinatorial effects, and to determine the translational therapeutic potential of this combination using primary patient samples and mouse models.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal