

Acute myeloid leukaemia (AML) is the most common acute leukaemia in adults, with an incidence of 3-5 cases per 100,000 individuals, while seen in less than 10% of acute leukaemia in children. Combined morphologic-phenotypic, cytogenetic and molecular results allow sub classification of the disease. The 2017 WHO classification recognizes nine chromosomal and molecular abnormalities that define specific AML subtypes, which account for 20-30% of the AML cases. In addition to disease classification, these genome profiles are used for risk stratification of AML into favorable, intermediate and poor risk prognostic groups. Routine genetic profiling on all newly diagnosed AML patients is the standard of care today, and its importance will likely continue to increase in the future.
MYLOTARG (gemtuzumab ozogamicin, a drug linked monoclonal antibody) in combination with chemotherapy offers a statistically significant improvement in median event-free survival versus chemotherapy alone (17.3 months and 9.5 months, respectively). It is however recommended only in AML with favourable or intermediate cytogenetic profile and treatment needs to start immediately for optimal response. Therefore there is a demand to identify the genome complexity of AML well below the current reporting times of 10 calendar days. Using standard banding techniques, an abnormal karyotype can be detected in 50 to 60 percent of patients with de novo AML (Grimwade et al., Blood 2010). It provides a global genome view albeit at a limited resolution of >10Mb but is a long skill dependent test, which takes over 150 hrs. While Fluorescence in situ hybridization (FISH) is a much faster highly sensitive assay for detecting both structural and numerical abnormalities it is limited to single target. In contrast chromosomal microarray analysis (CMA) allows assessment at very high resolution of genome wide imbalances and combined with FISH screen for fusion genes offers a speedy genetic evaluation of AML (Xu et al., Cancer Genetics, 2018).
Here we present a comparative study of AML presentation samples showing that genome profiling based on CMA and FISH offers an alternative to classical karyotyping for fast reliable genome assessment ready for use in routine diagnostic settings. Total of 171 presentation bone marrow adult AML samples were investigated using classical G banding analysis, fusion gene screening by FISH including CBFB /MYH11, RUNX1/RUNXT1, EVI (MECOM)/BA, MLL (KMT2A/BA), PML/RARA (Cytocell), confirmed by multiplex PCR assay Q30 (QuanDX) and CMA using oligonucleotide arrays (Agilent). Additional FISH and or Q30 to validate the CMA results were performed as needed.
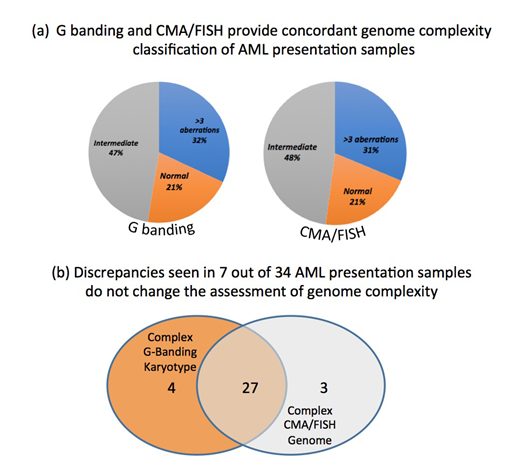
In 101 /171 samples, G banding data was compared with the genome profile based on combined CMA/fusion gene screened (FISH/Q30) data. The number of normal, intermediate (1-3 aberrations) and complex (> 4 aberrations) genomes identified by both approaches was very similar (fig 'a'). Both methods showed lack of aberration in 21 % of the samples, referred to as 'normal', 47% and 48% samples resp. had up to 3 genome changes ('intermediate'), while the remaining 31% and 32% resp. contained more than 4 numerical and/or structural aberrations.
Altogether 9 out of the 101 (8.9%) samples were found to have discrepant G banding/CMA results. However these discrepancies did not affect the genome classification (fig 'b'). For examples in the group of complex genomes 4 cases had aberrations detected by limited G banding, which were not confirmed by FISH or detected by CMA and likely reflect the restricted resolution of the chromosome analysis. In contrast the additional genome lesion detected by CMA were cryptic (below 10Mb in size) aberrations well beyond the resolution of the G banding. Among the most frequently detected cryptic changes are deletions of TET2 at 4q24, TP53 at 17p13 and RUNX1 at 21q11 gene regions. In addition, loss at the short arm of chromosome 3 and 12, long arms of chromosome 16 and 18, along with deletion the long arm of chromosome 20 were detected at 10% levels.
Similar features were detected in the genome profiling based on CMA and FISH of further 70 AML samples with failed G banding analysis, which further demonstrates that the CMA/FISH approach is readily applicable for genome evaluation of AML at presentation.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal