

Introduction: CXCL9 is a CXCR3 ligand that was shown to be implicated in the pathomechanism of cGVHD after allogeneic stem cell transplantation (alloSCT). In the present study, we screened single nucleotide polymorphisms (SNPs) inside the CXCL9 locus, and measured peritransplant CXCL9 serum levels kinetics to further determine the role of CXCL9 in cGVHD pathogenesis.
Patients and methods: Patients surviving at least 6 months after alloSCT with available DNA or sera were eligible for this study. 317 patients who were allografted at the University of Heidelberg and did not receive statin-based endothelial prophylaxis (SEP) constituted the no-SEP training cohort, 424 patients from Heidelberg who received SEP constituted the SEP cohort. The no-SEP validation cohort consisted of 211 patients who were allografted at an independent institution (Charite Berlin). Patients were retrospectively graded for cGVHD based on the NIH consensus criteria.
Prospectively collected serum samples taken once weekly before alloSCT until day+28 were analyzed for CXCL9 serum levels in n=132 / n=331 patients (no SEP/SEP resp.). CXCL9 serum levels dropped within two weeks after conditioning therapy and normalized until day+28. Minimal serum concentrations measured between days 0-14 and recovery serum values measured on day+28 were used to calculate the slope of CXCL9 recovery in the context of calcineurin inhibitors.
Four SNPs (rs4282209, rs3733236, rs2276885 and rs884304) in CXCL9 locus were selected based on linkage disequilibrium (LD) and tagging SNP approach. The SNPs were genotyped in recipient DNA and analyzed for association with severe cGVHD. Hazard ratios (HR) with 95% confidence interval (CI) were estimated using Cox regression. Multivariate analysis included age, diagnosis, matched vs. mismatched donor, sex of donor and recipient and usage of ATG as covariates.
Functionality of the regulatory SNP rs884304 was investigated using luciferase reporter assay in HEK293T cells. A total of 5 reporter constructs were generated with Quik-change site-directed mutagenesis kit (Invitrogen). In addition to the wildtype construct, three constructs carried variant alleles for each of the three SNPs, and one construct carried variant alleles for all the three SNPs. The constructs were transfected using lipofectamine 2000 reagent and incubated for 24h without treatment or treated with IFN- γ and/or cyclosporine (CsA) or tacrolimus. Cells were subsequently lysed and assayed for luciferase activity.
Results: Overall, 60 of 317 patients (19%) in the no-SEP training cohort, 54 of 424 patients (13%) in the SEP cohort, and 50 of 211 patients (24%) in the validation cohort developed at least one episode of severe cGVHD. Out of the four SNPs, only the AA/AG genotypes for the rs884304G>A polymorphism associated with significantly higher risk of severe cGVHD in the no-SEP training cohort (HR: 2.01, 95%CI 1.04-3.90, P=0.04). The effect was also reproduced in the no-SEP validation cohort (HR: 1.88, 95%CI 1.06-3.32, P=0.04). In contrast, no significant effect was observed for high risk genotypes in the SEP cohort (HR 0.77, 95% CI 0.41-1.46, P=0.43).
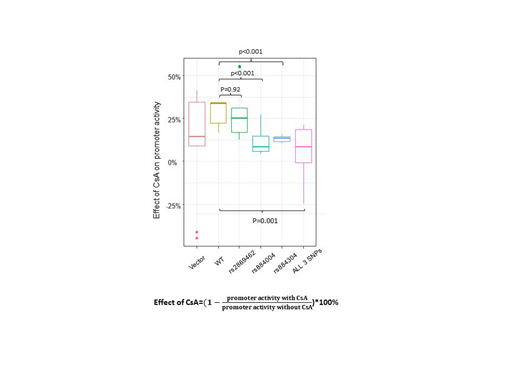
In reporter assays in the context of IFN- γ activation, significantly reduced suppressive effects of CsA on the promoter activity in the constructs with variant alleles of SNP rs884304 (P<0.001) and rs884004 (P<0.001) was observed compared to the wildtype construct (Figure 1).
The slope of CXCL9 serum level increase until day+28 significantly correlated with the incidence of severe cGVHD (every 1 unit, HR 1.007, 95%CI 1.001-1.014, P=0.03) and was higher in high risk genotypes of rs884304 (Kruskal-Wallis test p=0.02).
Conclusion: Rapidly rising CXCL9 serum levels after alloSCT are associated with an increased risk of developing severe cGVHD. CXCL9 SNPs rs884304 and rs884004 alter CXCL9 suppression in response to calcineurin inhibitors, representing one possible mechanism for enhanced CXCL9 expansion and subsequent cGVHD promotion.
Figure 1: Reduced suppressive effect of CsA on CXCL9 promoter activity with HR SNPs in the context of IFN- γ activation.
Dreger:AbbVie, AstraZeneca, Gilead, Janssen, Novartis, Riemser, Roche: Consultancy; AbbVie, Gilead, Novartis, Riemser, Roche: Speakers Bureau; Neovii, Riemser: Research Funding; MSD: Membership on an entity's Board of Directors or advisory committees, Other: Sponsoring of Symposia. Luft:JAZZ: Research Funding; Neovii: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal