

In this issue of Blood, 1 present an extensive in-depth genetic characterization of diagnostic, relapse, and remission samples from a cohort of 103 pediatric patients with acute lymphoblastic leukemia (ALL) treated according to the Shanghai Children’s Medical Center ALL-2005 frontline protocol. Together with data obtained from 208 serial bone marrow samples collected during ALL therapy, their work suggests that relapse in a fraction of childhood ALL patients is driven by chemotherapy-induced mutations, which impact therapy response.
Schematic overview of the differences between very early and early-late relapse in pediatric ALL. NGS, next-generation sequencing.
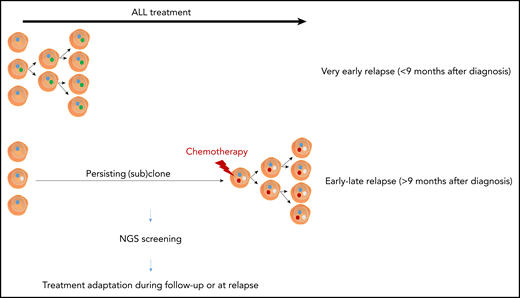
Schematic overview of the differences between very early and early-late relapse in pediatric ALL. NGS, next-generation sequencing.
Since the early days of cancer treatment with chemotherapy in the 1960s, the cure rate of childhood ALL has dramatically improved.2 Indeed, overall survival rates for pediatric ALL patients are currently well above 90% with contemporary treatment protocols that use minimal residual disease (MRD) measurements to guide treatment intensity.2 Nevertheless, disease relapse is still a major clinical problem, and the biology of relapsed disease as well as the molecular mechanisms that drive therapy resistance remain poorly understood.2
By means of whole-genome sequencing, Li et al identified a set of 12 mutations that were significantly enriched or exclusively present at relapse in a large and uniformly treated cohort of pediatric ALL. Of note, this patient population only included 16 diagnosis-remission-relapse trios from T-cell ALL (T-ALL) patients, with a bias toward tumor material obtained from TAL1, TAL2, LMO2, or LMO3 rearranged T-ALLs. Therefore, some of the results obtained in this study might not be readily transferable toward all genetic subtypes of T-ALL.3
Most of the relapse-associated genetic defects found in this work have previously been identified in relapsed ALL (TP53, NR3C1, NR3C2, CREBBP, WHSC1, NT5C2, PRPS1, PRPS2, MSH2, MSH6, and PMS2)4,5 and are thought to affect chemotherapy responses to key components of ALL therapy, such as steroids or thiopurines.6 However, in this study, Li et al also discovered that mutations and focal deletions in the folate metabolism gene FPGS exclusively occur in relapsed ALL and cause increased resistance to methotrexate, a core component of ALL consolidation therapy.
Interestingly, very early relapses (<9 months after diagnosis) harbored few of the relapse-specific mutations mentioned above, suggesting that they probably originate from a clone that already existed at diagnosis (see figure). This preexisting resistance model for very early ALL relapse was further supported by doubling time calculations based on serial MRD samples that were available from 19 B-cell ALL patient samples and publicly available datasets.5 Interestingly, these very early relapses mainly occurred in ALL cases harboring mixed-lineage leukemia or BCR-ABL rearrangements, aberrations notoriously associated with poor response to chemotherapy.2,7
In contrast to very early relapses, relapse-specific mutations were significantly enriched in cases arising later than 9 months after diagnosis, thereby pointing to potential on-treatment acquired resistance in a persisting clone (see figure). Indeed, direct involvement of the uncovered mutations to response to drugs used for ALL treatment (NR3C1/2, CREBBP, and WHSC1 for glucocorticoids; NT5C2, MSH2/6, PMS2, and PRPS1/2 for thiopurines; and FPGS for methotrexate) suggests that ALL disease progression can be directly affected by chemotherapy-induced mutations.
Although usually only a fraction of mutations occurring in cancer is considered as drivers and actionable therapeutic targets, the identification of the complete genetic landscape by whole-genome sequencing approaches can provide insights into the mutational processes that shape cancer genomes and serve as biomarkers as well as targets for therapeutic intervention.8 So far, 30 mutational cancer signatures (ie, unique combinations of mutations in primary cancers) have been identified and categorized in the COSMIC database. For about one-half of these mutational fingerprints, possible underlying molecular mechanisms have been suggested, for example, increased activity of APOBEC enzymes. This family of cytidine deaminases generates protein diversity by means of messenger RNA editing and plays a crucial role in B-cell receptor somatic hypermutation. Two of the 9 known mutational signatures identified in the ALL samples involved APOBEC and were detected at both diagnosis and relapse. Notably, the identification of such signatures can be therapeutically relevant, as exemplified by the APOBEC signature that can predict response toward checkpoint blockade immune therapy in non–small cell lung cancer.9 Therefore, relapsed ALL cases harboring the APOBEC signature might also be eligible for immune therapy.
Interestingly, 2 novel mutational signatures were uniquely detected in the dominant clone of >25% of ALL relapsed samples in this study, indicating that they were acquired early during the course of the treatment. Of note, in vitro exposure experiments, using the nontumorigenic epithelial cell line MCF10A, identified thioguanines as the causative agents of one of these signatures, which was responsible for at least a subset of the relapse-specific mutations mentioned above. Although the exact mechanisms by which thiopurines cause this mutational signature remain unknown, these findings have major clinical implications because they indicate that a fraction of ALL relapses could potentially be prevented in the future by adapting frontline ALL treatment strategies.
Altogether, the relapse-specific mutations as well as the mutational signatures reported in this study provide a causative explanation for a subset of ALL relapses and pave the way toward the identification of biomarkers that could identify therapy resistance early on during therapy or treatment follow-up. In addition, as >50% of relapsed ALL samples harbor at least 1 mutation potentially affecting therapy response, the mutational resistance profile of each refractory ALL patient should be taken into account to personally adapt the treatment schedule at relapse. Finally, in a subset of relapsed ALL samples, the genetic aberrations found in the tumor cells are not the clear culprits of resistance, suggesting that also other, potentially nongenetic factors, could mediate certain aspects of therapy resistance.10 Therefore, both genetic and nongenetic mechanisms of therapy resistance should eventually be taken into account to clinically manage relapsed ALL.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal