

In an unexpected transsignaling pathway, in this issue of Blood, 1 show that the toll-like receptor (Tlr)4 adaptor TIR-domain–containing adapter-inducing interferon-β (TRIF) redirects liver-derived high-mobility group box 1 (HMGB1) to activate myeloid cells to a highly procoagulant state in sepsis.
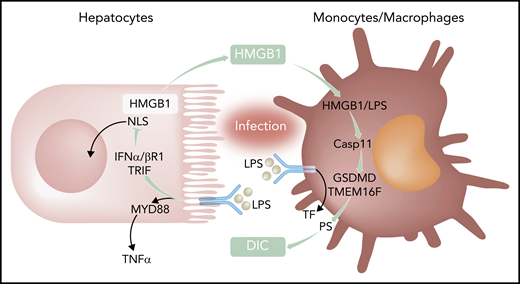
Activation of innate immunity by bacterial products leads to Tlr4-MyD88–dependent inflammation and induction of the coagulation initiator TF. Coupling of Tlr4 to TRIF-IFN-α/βR1 targets nuclear localization sequences (NLS) of HMGB1 and increases availability of cytosolic HMGB1 for secretion and extracellular functions. In macrophages, LPS delivered by HMGB1 triggers activation of the noncanonical caspase 11 (Casp11) for gasdermin D (GSDMD) and transmembrane protein 16F (TMEM16F)-mediated PS translocation and TF-dependent DIC. TNFα, tumor necrosis factor α.
Activation of innate immunity by bacterial products leads to Tlr4-MyD88–dependent inflammation and induction of the coagulation initiator TF. Coupling of Tlr4 to TRIF-IFN-α/βR1 targets nuclear localization sequences (NLS) of HMGB1 and increases availability of cytosolic HMGB1 for secretion and extracellular functions. In macrophages, LPS delivered by HMGB1 triggers activation of the noncanonical caspase 11 (Casp11) for gasdermin D (GSDMD) and transmembrane protein 16F (TMEM16F)-mediated PS translocation and TF-dependent DIC. TNFα, tumor necrosis factor α.
Gram-negative bacteria and lipopolysaccharide (LPS) triggering of Tlr4 are major inducers of inflammation and de novo synthesis of tissue factor (TF), the major coagulation initiator, in sepsis and other systemic inflammatory syndromes. TF expressed by monocytes and macrophages is crucial for the development of disseminated intravascular coagulation (DIC), which is responsible for much of the lethality of sepsis. In a mouse model of sepsis in which mice are challenged with LPS after nonlethal LPS priming, Yang et al found that Tlr4 adaptors play differential roles in inducing inflammation and DIC, as evidenced by markers of platelet consumption, intravascular fibrin deposition, and thrombin generation (see figure). Whereas canonical NF-κB signaling through myeloid differentiation primary response gene 88 (MyD88) was responsible for inflammatory tumor necrosis factor α production, only deletion of TRIF markedly attenuated the systemic coagulopathy. Deficiency of TRIF and downstream type 1 interferon signaling through interferon-α/β receptor I (IFN-α/βR1) also diminished DIC in the cecum ligation and puncture model, emphasizing the relevance of this newly described signaling mechanism for bacterial sepsis.
These authors have previously shown that type 1 interferon signaling induces hyperacetylation of HMGB1. Hyperacetylation inactivates the nuclear location sequences and thereby increases HMGB1 cytoplasmic accumulation and availability for release into the extracellular space, where HMGB1 has sepsis-promoting properties. Consistent with a functional role for this pathway in sepsis, HMGB1 levels were reduced in TRIF−/− and IFN-α/βR1−/−, but not MyD88−/− mice following challenge. Mice with cell type–specific deletions of HMGB1 excluded myeloid cells and implicated hepatocytes as the primary source for HMBG1 circulating in the blood of septic mice. Platelet-derived HMGB1 has previously been shown to promote thrombosis,2 but neither hematopoietic cell nor platelet-specific deletion of HMGB1 abrogated DIC in sepsis. Conversely, although interrupting TRIF-IFN-α/βR1-HMGB1 signaling was sepsis protective, IFN-α/βR1−/− mice did not have a defect in arterial thrombosis, demonstrating the selective significance of the identified signaling mechanism for DIC associated with infectious diseases.
Although the authors confirmed prior data that monocyte/macrophage–derived TF is crucial for the coagulopathy in sepsis, interruption of the TRIF-IFN-α/βR1-HMGB1 axis did not measurably reduce the elevated TF messenger RNA and protein levels in organs of challenged mice. Data with purified macrophages provided compelling evidence that HMGB1 is directly responsible for increasing cell surface availability of phosphatidylserine (PS), which, thereby, alters the procoagulant activity of TF (see figure). These results are in line with expanding evidence that the activity of monocyte-expressed TF in thrombosis3 and autoimmune disease4 is regulated by rapid posttranslational mechanisms. Macrophages express TF in a cryptic form on the cell surface, but cellular stress signals, such as ATP triggering the purinergic P2X7 receptor, can rapidly activate TF by increasing extracellular PS coupled to inflammasome/caspase 1–dependent release of highly procoagulant extracellular vesicles.5 TF can also be activated by an inflammasome-caspase 11–dependent pathway leading to gasdermin D–dependent PS exposure and pyroptoptosis.6 HMGB1 activates this noncanonical inflammasome pathway by delivering LPS for cytosolic activation of caspase 11, gasdermin D, and the PS scramblase transmembrane protein 16F. As shown in this paper, the resulting externalization of PS was required for macrophage TF procoagulant activity and, accordingly, inhibition of PS reduces DIC in sepsis challenged mice.
DIC is typically viewed as a failure of critical anticoagulant mechanisms due to reciprocal amplification of inflammation and coagulation. The current study provides conceptually new insight that a specific arm of the innate immune response connects tissue stress to the posttranslational activation of TF on immune cells. Complement factor (C) 3, which is part of the other major plasmatic innate defense pathway, is also linked to TF activation and participates in thrombosis and autoimmune signaling by supporting thiol isomerase–dependent conformational changes in TF.3,4 Blockade of C3 prevents the coagulopathy in sepsis,7 and HMGB1 can activate C3 in sterile inflammation.8 It is therefore conceivable that the identified role of extracellular HMGB1 in creating a procoagulant environment on the cell surface extends to complement-mediated effects that allosterically activate TF.
Not only are TRIF-IFN-α/βR1 signaling and coagulation connected in this link promoting DIC, but also, intriguingly, the TF coagulation initiation signaling complex activating protease activated receptor 2 directly controls Tlr4-TRIF responses in sepsis.9 These signaling events are further regulated in innate immune cells by the sepsis-protective anticoagulant pathway.10 The new connection of DIC induction uncovered here may, therefore, prove to be part of a broader crosstalk between coagulation and immunity in infectious diseases.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal