

Key Points
HDIVig appears to be an effective treatment for nonsevere forms of scleromyxedema. Maintenance therapy is required to avoid recurrence.
Chemotherapies targeting the MG-secreting plasma cell clone seem to be useful for refractory and severe forms of this disease.

Abstract
Scleromyxedema is a rare skin and systemic mucinosis that is usually associated with monoclonal gammopathy (MG). In this French multicenter retrospective study of 33 patients, we investigated the clinical and therapeutic features of MG-associated scleromyxedema. Skin molecular signatures were analyzed using a transcriptomic approach. Skin symptoms included papular eruptions (100%), sclerodermoid features (91%), and leonine facies (39%). MG involved an immunoglobulin G isotype in all patients, with a predominant λ light chain (73%). Associated hematologic malignancies were diagnosed in 4 of 33 patients (12%) (smoldering myeloma, n = 2; chronic lymphoid leukemia, n = 1; and refractory cytopenia with multilineage dysplasia, n = 1). Carpal tunnel syndrome (33%), arthralgia (25%), and dermato-neuro syndrome (DNS) (18%) were the most common systemic complications. One patient with mucinous cardiopathy died of acute heart failure. High-dose IV immunoglobulin (HDIVig), alone or in combination with steroids, appeared to be quite effective in nonsevere cases (clinical complete response achieved in 13/31 patients). Plasma cell–directed therapies using lenalidomide and/or bortezomib with dexamethasone and HDIVig led to a significant improvement in severe cases (HDIVig refractory or cases with central nervous system or cardiac involvement). The emergency treatment of DNS with combined plasmapheresis, HDIVig, and high-dose corticosteroids induced the complete remission of neurological symptoms in 4 of 5 patients. Quantitative reverse-transcriptase polymerase chain reaction analysis of 6 scleromyxedema skin samples showed significantly higher profibrotic pathway levels (transforming growth factor β and collagen-1) than in healthy skin. Prospective studies targeting plasma cell clones and/or fibrotic pathways are warranted for long-term scleromyxedema management.
Introduction
Scleromyxedema is a rare systemic mucinosis characterized by generalized papular and sclerodermoid cutaneous eruptions that is usually associated with monoclonal gammopathy (MG) that involves an immunoglobulin G (IgG) isotype with slow electrophoretic mobility.1 Scleromyxedema most often results from a small secretory plasma cell clone (commonly referred to as “MG of undetermined significance”; MGUS) that may be associated with severe organ damage.2 Scleromyxedema could be part of MG of clinical significance,3 but it may occur more rarely in the setting of MG associated with B-cell and plasma cell malignancies, such as myeloma and B-cell lymphomas.4,5 Various types of extracutaneous involvement have been described in scleromyxedema, particularly neurological, joint, gastrointestinal, and heart impairments.4 Mortality of scleromyxedema remains high, primarily as a result of treatment toxicity and dermato-neuro syndrome (DNS), a severe acute encephalopathy that presents as epileptic seizures and coma. The efficacy of high-dose IV immunoglobulin (HDIVig) for scleromyxedema skin symptoms has been described,4,6,7 but no report has been published on the management of severe and refractory forms of scleromyxedema. Additionally, only limited data are available concerning the benefit of plasma cell–directed therapies, particularly in MGUS-associated scleromyxedema. The physiopathology of scleromyxedema remains largely unknown. A small study showed that patient serum (but not purified monoclonal protein alone) could induce fibroblast proliferation in vitro, suggesting that there may be an unknown relevant factor in the serum.8,9 Skin molecular signatures of scleromyxedema have never been investigated.
The main objective of this retrospective study was to gain new insights into the severe visceral organ involvement of scleromyxedema and its therapeutic management while studying the largest patient cohort to date, with a particular focus on plasma cell–directed therapies. Skin molecular pathways of fibroblast activation, mucin production, inflammation, and fibrosis were analyzed.
Patients and methods
Patient inclusion and data collection
We conducted a multicenter nationwide study in France that included patients with scleromyxedema. Patients had to fulfill ≥3 of the 4 Rongioletti and Rebora criteria: (1) papular eruption, (2) mucin deposition, fibroblast proliferation, and fibrosis on skin histology, (3) MG, and (4) the absence of thyroid disease. MGUS was diagnosed in the absence of hematological diseases, according to the International Myeloma Working Group criteria.10 Thirty-three scleromyxedema patients who were diagnosed between January of 1999 and June of 2018 in dermatology (n = 18), internal medicine (n = 3), hematology (n = 3), and rheumatology (n = 1) departments were included.
Disease severity and treatment response definitions
Severe vs nonsevere scleromyxedema cases were defined as cases with or without DNS and/or mucinous cardiac involvement, respectively. Clinical response was defined as complete response (CR; complete clinical improvement from baseline), partial response (PR; a clinical improvement > 25% from baseline), or no response (NR; stability or worsening). The different lines of therapy (thalidomide, corticosteroids alone, acitretin, melphalan + dexamethasone, and methotrexate) were analyzed separately for each patient. Hematological response was defined as CR (disappearance of MG and negative immunofixation), PR (decrease in serum MG levels > 50%), MR (decrease in serum MG levels > 25%), or NR (no change or increase in serum MG levels).
Skin transcriptomic analysis
Total RNA was extracted from 5 20-μm-thick paraffin skin sections using an RNA extraction kit and an FFPE RNeasy Kit (QIAGEN, Valencia, CA), as described previously.11 After elution on RNeasy MinElute spin columns, RNA was transcribed into complementary DNA by reverse transcriptase using Reverse Transcription Master Mix (Fluidigm); oligo dTs were used as the primer for complementary DNA synthesis (primer sequences are described in supplemental Data 1, available on the Blood Web site). Quantitative polymerase chain reaction (PCR) was performed using Applied Biosystems Power SYBR Green PCR Master Mix (Invitrogen). The quantification of the expression of each gene was performed using the comparative cycle threshold (Ct) method, according to the manufacturer’s instructions (Applied Biosystems). An average of the duplicate reaction results was normalized to the reference β-2 microglobulin for each gene (ΔCt = Ct sample − Ct β-2 microglobulin). ΔCt values are expressed as arbitrary units, and the results are expressed as the mean ratios of scleromyxedema ΔCt values/normal skin ΔCt values for each gene. Mean ratios were compared using the Mann-Whitney test. Healthy skin from plastic surgery patients was used as control skin. Skin samples were acquired after obtaining informed consent from patients according to our local ethics rules (CPP, Paris X).
Statistical analysis
The data are expressed as medians with ranges, means with standard deviations (SDs), and numbers with frequencies. The Mann-Whitney test was used to compare continuous variables. Probability of overall survival of all 33 patients and overall survival without DNS or specific mucinous cardiomyopathy were evaluated using the Kaplan-Meier method, considering the probability of event over time. All tests were 2 sided, and a value of P < .05 was considered statistically significant. Statistical analysis was performed using GraphPad Prism 5.
Results
Patient characteristics
The demographic, clinical, biological, and histological characteristics of the 33 patients are summarized in Table 1. The mean age at diagnosis was 56.3 ± 13.6 years (range, 28-78 years), with a male/female (M/F) ratio of 1.06. The median follow-up was 4.3 ± 2.39 years (range, 0.5-13 years).
Characteristics of 33 scleromyxedema patients
| Results | |
|---|---|
| Patients | |
| Sex ratio (M/F) | 1.06 (17/16) |
| Age, mean ± SD, y | 55.4 ± 13.6 |
| Follow-up, mean ± SD, y | 4.3 ± 2.39 |
| Death | 1 (3) |
| Cutaneous manifestations | 33 (100) |
| Papular eruption/pruritus | 33 (100)/18 (54) |
| Sclerodermoid eruption/leonine facies | 30 (90)/13 (39) |
| Depilation/alopecia | 10/22 (45)/4 (12) |
| Purpura/livedo racemosa/cutis laxa–like syndrome/skin necrosis | 1 (3)/3 (9)/3 (9)/1 (3) |
| Extracutaneous manifestations | 22 (67) |
| Hematological involvement | 33 (100) |
| MG/MGUS/hematologic malignancy | 33 (100)/30 (91)/4 (12) |
| Neurological involvement | 18 (54) |
| Central nervous system involvement/DNS | 9 (27)/6 (18) |
| Carpal tunnel syndrome | 12 (36) |
| Sensitive polyneuropathy | 1 (3) |
| Cardiac and vascular involvement | 5 (15) |
| Ischemic/mucinous cardiopathy | 3 (9)/2 (6) |
| Arterial hypertension | 11 (33) |
| Pulmonary involvement | 3 (9) |
| Obstructive syndrome | 3 (9) |
| Interstitial pneumopathy | 1 (3) |
| Musculoskeletal involvement | 9 (27) |
| Myositis/myalgia | 2 (6)/2 (6) |
| Arthralgia/arthritis | 9 (27)/1 (3) |
| Gastrointestinal involvement (dysphagia) | 2 (6) |
| Head and neck involvement (dysphonia) | 1 (3) |
| Ophthalmic (retrobulbar optic neuritis) | 1 (3) |
| Laboratory findings | |
| MG/biclonal gammopathy | 33 (100)/1 (3) |
| Serum IgG level, g/L | 4.5 ± 3 |
| κ or λ light chain in monoclonal Ig | 10/33 (29) or 24/33 (73) |
| Skin histological findings | 33 (100) |
| Dermal mucinosis/fibroblast proliferation/fibrosis | 33 (100) |
| Granuloma annulare form: histiocytic infiltrate (CD68+ and CD163+) | 10 (27) |
| Results | |
|---|---|
| Patients | |
| Sex ratio (M/F) | 1.06 (17/16) |
| Age, mean ± SD, y | 55.4 ± 13.6 |
| Follow-up, mean ± SD, y | 4.3 ± 2.39 |
| Death | 1 (3) |
| Cutaneous manifestations | 33 (100) |
| Papular eruption/pruritus | 33 (100)/18 (54) |
| Sclerodermoid eruption/leonine facies | 30 (90)/13 (39) |
| Depilation/alopecia | 10/22 (45)/4 (12) |
| Purpura/livedo racemosa/cutis laxa–like syndrome/skin necrosis | 1 (3)/3 (9)/3 (9)/1 (3) |
| Extracutaneous manifestations | 22 (67) |
| Hematological involvement | 33 (100) |
| MG/MGUS/hematologic malignancy | 33 (100)/30 (91)/4 (12) |
| Neurological involvement | 18 (54) |
| Central nervous system involvement/DNS | 9 (27)/6 (18) |
| Carpal tunnel syndrome | 12 (36) |
| Sensitive polyneuropathy | 1 (3) |
| Cardiac and vascular involvement | 5 (15) |
| Ischemic/mucinous cardiopathy | 3 (9)/2 (6) |
| Arterial hypertension | 11 (33) |
| Pulmonary involvement | 3 (9) |
| Obstructive syndrome | 3 (9) |
| Interstitial pneumopathy | 1 (3) |
| Musculoskeletal involvement | 9 (27) |
| Myositis/myalgia | 2 (6)/2 (6) |
| Arthralgia/arthritis | 9 (27)/1 (3) |
| Gastrointestinal involvement (dysphagia) | 2 (6) |
| Head and neck involvement (dysphonia) | 1 (3) |
| Ophthalmic (retrobulbar optic neuritis) | 1 (3) |
| Laboratory findings | |
| MG/biclonal gammopathy | 33 (100)/1 (3) |
| Serum IgG level, g/L | 4.5 ± 3 |
| κ or λ light chain in monoclonal Ig | 10/33 (29) or 24/33 (73) |
| Skin histological findings | 33 (100) |
| Dermal mucinosis/fibroblast proliferation/fibrosis | 33 (100) |
| Granuloma annulare form: histiocytic infiltrate (CD68+ and CD163+) | 10 (27) |
Unless otherwise noted, data are n (%).
The clinical skin features included papular eruptions (n = 33; 100%) (Figure 1A-C), sclerodermatous features (n = 30; 91%), leonine facies (n = 13; 39%) (Figure 1B), pruritus (n = 18; 54%), livedo racemosa (n = 3; 9%) (Figure 1D), and lax skin that was consistent with cutis laxa (n = 3; 9%). The histological skin features included mucin deposits, fibroblastic proliferation, and fibrosis in all patients. A granuloma annulare–like pattern with histiocytic CD68+ or CD163+ infiltrate was found in 10 patients (30%) (supplemental Data 1). Extracutaneous involvement occurred in 21 patients (64%), which primarily involved carpal tunnel syndrome (n = 11; 33%), arthralgias (n = 9; 27%), and dysphagia (n = 2; 6%). Five patients (15%) had a cardiac event during follow-up: ischemia (n = 3), an event that was related to specific heart scleromyxedema involvement (n = 2), mucin deposition with acute heart failure and death (n = 1), and right ventricular dilatation and tricuspid valvulopathy (n = 1) (Table 1).
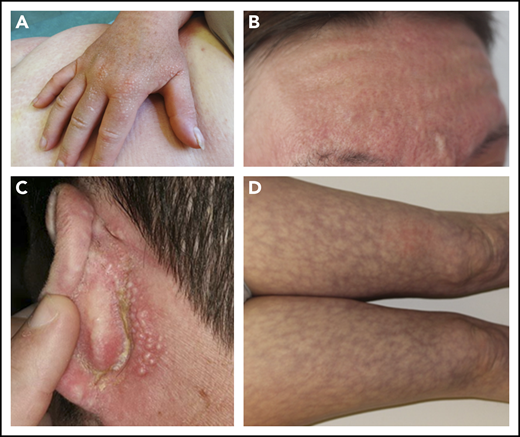
Skin features in scleromyxedema. Papules on hands (A), glabella (B), and retroauricular area (C).(D) Livedo racemosa.
Skin features in scleromyxedema. Papules on hands (A), glabella (B), and retroauricular area (C).(D) Livedo racemosa.
Six patients had a typical DNS, which occurred in 5 patients >1 year after the diagnosis (Table 2). Epileptic seizures and coma were preceded by fever (n = 3), flu-like syndromes (n = 4), and various neurological symptoms, such as dysarthria (n = 2), tetrapyramidal syndrome (n = 1), cerebellar ataxia (n = 2), hemiparesis (n = 2), chorea (n = 1), psychosis (n = 1), aphasia (n = 1), and visual hallucinations (n = 1). Orotracheal intubation was performed in 4 patients. Cerebrospinal fluid examinations (n = 5) and brain imaging with cerebral computed tomography angiography (n = 2) or magnetic resonance imaging (MRI) (n = 5) were normal in all patients. The MG isotype was IgG in all patients (100%), with λ light chain in 24 patients (73%). Four patients (12%) had an associated hematologic malignancy: 2 had smoldering myeloma, 1 had chronic lymphocytic leukemia, and 1 had refractory cytopenia with multilineage dysplasia. Bone marrow aspiration was performed in 30 patients and was strictly normal in 23 patients (76%). Four patients displayed <10% plasma cells with cytological abnormalities, and 2 patients had >10% plasma cells with cytological abnormalities. One patient with cytopenia was diagnosed with myelodysplasia that was unrelated to the plasma cell disorder. One patient had monoclonal B lymphocytosis. Slow electrophoretic mobility was observed in 8 of the 9 sera samples analyzed.
Characteristics of the 6 patients with DNS
| Patients | Age, y | Sex | Characteristics | Chronology | Previous treatment | Symptoms | Explorations | Treatment | Outcome/follow-up |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 28 | F | IgG κ, livedo | 1 y after diagnosis | HDIVig (stopped 3 mo before) | Flu-like syndrome, confusion, hemiparesis, epilepsy, and SE | cMRI: N. CSF: N. | ET: OTI, AEs, plasmapheresis, and HDIVig → slow CR. MT: lenalidomide + dexamethasone + HDIVig → CR. | Complete remission/1 y |
| 2 | 42 | M | IgG λ, carpal tunnel syndrome, smoldering myeloma | 5 y after diagnosis | HDIVig (spaced >6 wk apart) | Flu-like syndrome, fever, confusion, coma, and SE | cMRI: N. CSF: slight protein increase. | ET: OTI, AEs, plasmapheresis and HDIVig + CTs = CR. MT: lenalidomide + dexamethasone + HDIVig → CR. | Complete remission/2 y |
| 3 | 44 | M | IgG λ/GAL | 4 y after diagnosis | HDIVig | Flu-like syndrome, asthenia, fever, chorea, coma, epilepsy, and SE | cCT: N. | ET: OTI, AEs, plasmapheresis, HDIVIg → CR. MT: HDIVig → CR. | Complete remission/1 y |
| 4 | 68 | F | IgG λ/ GAL | 2 y before diagnosis (MGUS IgG λ) | — | Flu-like syndrome, fever, dysarthria, cerebellar syndrome, tetrapyramidal syndrome, coma, and SE | cMRI: N. EEG: N. CSF: N. | ET: OTI, AEs → PR. MT: HDIVig → CR. | Reduced psychomotor responses/4 y (complete skin response) |
| 5 | 57 | F | IgG λ and epilepsy ×3 (5 y before), purpura, skin necrosis, and vasculitis/GAL | 2 y after the first symptoms | — | Language dysfunction, epilepsy, and SE | cMRI: N, CSF: N, EEG: postcritical epilepsy. | ET: OTI, AEs, plasmapheresis, pulse methylprednisolone → CR. MT: bortezomib + dexamethasone. | Complete remission and epilepsy/2 y |
| 6 | 51 | M | IgG κ | 1 y after diagnosis (×5 episodes) | HDIVig | Confusion, asthenia, rotatory vertigo, convulsion, hemiparesis, hallucination psychosis, dysarthria, and SE | MRI: N ×3. cCT: N ×2. CSF: N ×5. EEG ×5: postcritical epilepsy. | ET: OTI, AEs → PR. MT: plasmapheresis and HDIVig = NR. Bortezomib/cyclophosphamide/dexamethasone and HDIVig → NR and relapse. Lenalidomide + bortezomib + dexamethasone → CR. | Complete remission (after the last episode) (after 5)/2 y (relapse after 2 y) |
| Patients | Age, y | Sex | Characteristics | Chronology | Previous treatment | Symptoms | Explorations | Treatment | Outcome/follow-up |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 28 | F | IgG κ, livedo | 1 y after diagnosis | HDIVig (stopped 3 mo before) | Flu-like syndrome, confusion, hemiparesis, epilepsy, and SE | cMRI: N. CSF: N. | ET: OTI, AEs, plasmapheresis, and HDIVig → slow CR. MT: lenalidomide + dexamethasone + HDIVig → CR. | Complete remission/1 y |
| 2 | 42 | M | IgG λ, carpal tunnel syndrome, smoldering myeloma | 5 y after diagnosis | HDIVig (spaced >6 wk apart) | Flu-like syndrome, fever, confusion, coma, and SE | cMRI: N. CSF: slight protein increase. | ET: OTI, AEs, plasmapheresis and HDIVig + CTs = CR. MT: lenalidomide + dexamethasone + HDIVig → CR. | Complete remission/2 y |
| 3 | 44 | M | IgG λ/GAL | 4 y after diagnosis | HDIVig | Flu-like syndrome, asthenia, fever, chorea, coma, epilepsy, and SE | cCT: N. | ET: OTI, AEs, plasmapheresis, HDIVIg → CR. MT: HDIVig → CR. | Complete remission/1 y |
| 4 | 68 | F | IgG λ/ GAL | 2 y before diagnosis (MGUS IgG λ) | — | Flu-like syndrome, fever, dysarthria, cerebellar syndrome, tetrapyramidal syndrome, coma, and SE | cMRI: N. EEG: N. CSF: N. | ET: OTI, AEs → PR. MT: HDIVig → CR. | Reduced psychomotor responses/4 y (complete skin response) |
| 5 | 57 | F | IgG λ and epilepsy ×3 (5 y before), purpura, skin necrosis, and vasculitis/GAL | 2 y after the first symptoms | — | Language dysfunction, epilepsy, and SE | cMRI: N, CSF: N, EEG: postcritical epilepsy. | ET: OTI, AEs, plasmapheresis, pulse methylprednisolone → CR. MT: bortezomib + dexamethasone. | Complete remission and epilepsy/2 y |
| 6 | 51 | M | IgG κ | 1 y after diagnosis (×5 episodes) | HDIVig | Confusion, asthenia, rotatory vertigo, convulsion, hemiparesis, hallucination psychosis, dysarthria, and SE | MRI: N ×3. cCT: N ×2. CSF: N ×5. EEG ×5: postcritical epilepsy. | ET: OTI, AEs → PR. MT: plasmapheresis and HDIVig = NR. Bortezomib/cyclophosphamide/dexamethasone and HDIVig → NR and relapse. Lenalidomide + bortezomib + dexamethasone → CR. | Complete remission (after the last episode) (after 5)/2 y (relapse after 2 y) |
AE, antiepileptic; cCT, cerebral computed tomography; cMRI, cerebral MRI; CSF, cerebrospinal fluid; CT, corticosteroid; EEG, electroencephalography; ET, emergency treatment; GAL, granuloma annulare–like histology; MT, maintenance treatment; N, normal; OTI, orotracheal intubation; SE, status epilepticus.
Skin transcriptomic analysis
Quantitative reverse-transcriptase PCR analysis of genes involved in fibroblast activation (α-smooth muscle actin), collagen production (transforming growth factor β [TGF-β] and collagen 1a), mucin production (CHSY1), elastic tissue production (elastin), and autoinflammatory pathway activation (CD68, interleukin-1βa [IL-1βa], IL-8, and IL-10) were compared in healthy (n = 6) and scleromyxedema skin samples (n = 6) (supplemental Data 2). The mean messenger RNA (mRNA) levels of TGF-β and collagen 1a were significantly higher in the skin samples from patients with scleromyxedema than in the skin from healthy donors (P = .05 and P = .004, respectively). IL-8 and IL-10 levels were also significantly increased in scleromyxedema samples (P = .02 and P = .05). α-Smooth muscle actin and elastin mRNA levels tended to be significantly higher in the skin of scleromyxedema patients than in the skin of normal patients, but the difference did not reach statistical significance (P = .1). The mRNA levels of CHSY1 and IL-1β were not significantly different.
Treatments
Nonsevere scleromyxedema
The first-, second-, and third-line therapies used in 32 patients with nonsevere scleromyxedema are summarized in Table 3. HDIVig (2 g/kg monthly for the first 6 months) was used as a first- or second-line therapy and was delivered for a median of 16.01 ± 9.04 months (range, 1.01-68.9 months). Clinical CR was achieved in 13 of 31 patients (42%) with HDIVig treatment, whereas CR was not observed in patients who were treated with thalidomide (100 mg/d; 4 patients: 2 PR and 2 NR) or lenalidomide (25 mg/d, 21/28 days; 2 patients: 1 PR and 1 NR). Corticosteroids alone (0.75 mg/kg per day of equivalent prednisone; 3 patients: 1 CR and 2 NR), acitretin (35 mg/d; 2 patients: 2 NR), melphalan + dexamethasone (10 mg/m2 by mouth on days 1-4 + 40 mg on days 1-4; 2 patients: 1 PR and 1 NR), and methotrexate (15 mg/wk; 1 patient: 1 NR) were frequently ineffective.
Management of nonsevere scleromyxedema
| Lines of therapy | Treatment | Mean dose | Mean follow-up duration, mo | Response, n | Failure, n (%) | DNS/cardiac injury or death, n | Severe side effects (≥grade 3-4), n |
|---|---|---|---|---|---|---|---|
| First line (n = 32) | HDIVig alone (n = 15) | 2 g/kg/m | 21 | 10 CR/5 PR | 2 (13) | 4 | 1 thrombosis |
| HDIVig + CTs (n = 5) | 2 g/kg/mo + 1 mg/kg/d | 10.2 | 5 PR | 1 (20) | 0 | 0 | |
| Thalidomide (n = 3) | 100 mg/d | 33.2 | 1 PR/2 NR | 2 (66) | 0 | 1 neuropathy | |
| Lenalidomide (n = 1) | 25 mg/d (21 d/28) | 11 | 1 NR | 1 (100) | 0 | 0 | |
| Acitretin (n = 2) | 35 mg/d | 8 | 2 NR | 2 (100) | 0 | 0 | |
| CTs alone* (n = 3) | 0.75 mg/kg/d | 6 | 1 CR/2 NR | 0 | 0 | 0 | |
| Melphalan + dexamethasone (n = 2) | 10 mg/m2 PO on days 1-4 + 40 mg on days 1-4 | 0.1 | 1 PR/1 NR | 2 (100) | 0 | 1 severe sepsis | |
| Methotrexate (n = 1) | 15 mg/wk | 3 | 1 NR | 1 (100) | 0 | 0 | |
| Second line (n = 11) | HDIVig (n = 6) | 2 g/kg/mo | 24 | 2 CR/4 PR | 1 (17) | 1 | 0 |
| HDIVig + CTs (n = 3) | 2 g/kg/mo + 0.3 mg/kg/d | 13 | 1 CR /2 PR | 0 | 0 | 0 | |
| Thalidomide + CTs (n = 1) | 100 mg/d + 0.25 mg/kg/d | 1 | 1 PR | 1 (100) | 0 | 1 neuropathy | |
| Lenalidomide (n = 1) | 25 mg/d (21 d/28) | 21 | 1 PR | 1 (100) | 0 | 0 | |
| Third line and more (n = 9) | HDIVig (n = 4) | 2 g/kg/mo | 28 | 4 PR | 0 | 0 | 0 |
| HDIVig + CT (n = 2) | 2 g/kg/mo + 0.5 mg/kg/d | 22.5 | 1 CR/1 PR | 0 | 0 | 0 | |
| HDIVig + lenalidomide (n = 1) | 2 g/kg/mo + 25 mg/d (21 d/28) | 16 | 1 CR | 0 | 0 | 0 | |
| HDIVig + thalidomide (n = 1) | 2 g/kg/mo + 100 mg/d | 3 | 1 PR | 0 | 0 | 0 | |
| Lenalidomide (n = 1) | 25 mg/d (21 d/28) | 4.73 | 1 PR | 0 | 0 | 0 |
| Lines of therapy | Treatment | Mean dose | Mean follow-up duration, mo | Response, n | Failure, n (%) | DNS/cardiac injury or death, n | Severe side effects (≥grade 3-4), n |
|---|---|---|---|---|---|---|---|
| First line (n = 32) | HDIVig alone (n = 15) | 2 g/kg/m | 21 | 10 CR/5 PR | 2 (13) | 4 | 1 thrombosis |
| HDIVig + CTs (n = 5) | 2 g/kg/mo + 1 mg/kg/d | 10.2 | 5 PR | 1 (20) | 0 | 0 | |
| Thalidomide (n = 3) | 100 mg/d | 33.2 | 1 PR/2 NR | 2 (66) | 0 | 1 neuropathy | |
| Lenalidomide (n = 1) | 25 mg/d (21 d/28) | 11 | 1 NR | 1 (100) | 0 | 0 | |
| Acitretin (n = 2) | 35 mg/d | 8 | 2 NR | 2 (100) | 0 | 0 | |
| CTs alone* (n = 3) | 0.75 mg/kg/d | 6 | 1 CR/2 NR | 0 | 0 | 0 | |
| Melphalan + dexamethasone (n = 2) | 10 mg/m2 PO on days 1-4 + 40 mg on days 1-4 | 0.1 | 1 PR/1 NR | 2 (100) | 0 | 1 severe sepsis | |
| Methotrexate (n = 1) | 15 mg/wk | 3 | 1 NR | 1 (100) | 0 | 0 | |
| Second line (n = 11) | HDIVig (n = 6) | 2 g/kg/mo | 24 | 2 CR/4 PR | 1 (17) | 1 | 0 |
| HDIVig + CTs (n = 3) | 2 g/kg/mo + 0.3 mg/kg/d | 13 | 1 CR /2 PR | 0 | 0 | 0 | |
| Thalidomide + CTs (n = 1) | 100 mg/d + 0.25 mg/kg/d | 1 | 1 PR | 1 (100) | 0 | 1 neuropathy | |
| Lenalidomide (n = 1) | 25 mg/d (21 d/28) | 21 | 1 PR | 1 (100) | 0 | 0 | |
| Third line and more (n = 9) | HDIVig (n = 4) | 2 g/kg/mo | 28 | 4 PR | 0 | 0 | 0 |
| HDIVig + CT (n = 2) | 2 g/kg/mo + 0.5 mg/kg/d | 22.5 | 1 CR/1 PR | 0 | 0 | 0 | |
| HDIVig + lenalidomide (n = 1) | 2 g/kg/mo + 25 mg/d (21 d/28) | 16 | 1 CR | 0 | 0 | 0 | |
| HDIVig + thalidomide (n = 1) | 2 g/kg/mo + 100 mg/d | 3 | 1 PR | 0 | 0 | 0 | |
| Lenalidomide (n = 1) | 25 mg/d (21 d/28) | 4.73 | 1 PR | 0 | 0 | 0 |
PO, by mouth.
Prednisone equivalent dose.
After HDIVig discontinuation (>3 months), relapse occurred in 7 of 8 patients, with a median relapse time of 13 months (range, 1-24 months). In patients who had previously achieved a primary CR, DNS occurred in 4 patients during HDIVig maintenance treatment.
The most common adverse side effects of HDIVig were headaches (15%), transient arterial hypertension (10%), chest pain (6%), and venous thrombosis (7%). Two patients treated with thalidomide developed grade 3 sensitive polyneuropathy.
Severe or HDIVig-refractory scleromyxedema
Seven patients with severe or HDIVig-refractory scleromyxedema were treated with plasma cell–directed therapies. Among these 7 patients, the combinations of lenalidomide, dexamethasone, and HDIVig (3 clinical PRs and 3 hematological CRs) and bortezomib, dexamethasone, and HDIVig (1 clinical CR and 1 hematological CR) resulted in a sustained response (Table 4).
Characteristics of 7 severe or HDIVig-refractory scleromyxedema patients treated with plasma cell–directed therapies
| Patient no. | Age, y | Sex | Characteristics | MG, level; hemopathy | Previous treatment | Status | Treatment | Duration/protocol | Clinical response | Hematologic response |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 65 | F | PE, myalgia, and arthralgia | κ, 4 g/L; MGUS | HDIVig | HDIVig-refractory disease | Bortezomib + cyclophosphamide + dexamethasone and HDIVig | 4 mo* | PR | CR (IF negative) |
| 2 | 61 | F | PE, CTS, and arthralgia | λ, 4 g/L; MGUS | HDIVig | HDIVig- refractory disease | Bortezomib + melphalan + dexamethasone | 1 mo† | NR, death | NR |
| 3 | 50 | M | PE, recurrent DNS, and CTS | κ, 3.9 g/L; MGUS | HDIVig | HDIVig-refractory and severe disease | Bortezomib + cyclophosphamide + dexamethasone | 2 mo* | NR | PR |
| Bortezomib + lenalidomide + dexamethasone | 5 mo‡ | PR (almost complete) | CR (IF negative) | |||||||
| 4 | 43 | M | PE, DNS, and CTS | λ, 4.5 g/L; smoldering myeloma | HDIVig + CT | HDIVig-refractory disease | Lenalidomide + dexamethasone + HDIVig | 6 mo§ | PR (almost complete) | CR (IF negative) |
| 5 | 63 | F | PE, DNS, epilepsy, and ischemic cardiopathy | κ, 11 g/L; MGUS | — | Severe disease | Bortezomib + dexamethasone | 4 mo¶ | CR | CR (IF negative) |
| 6 | 29 | F | PE, DNS, livedo, and GAL | λ, 7 g/L; MGUS | HDIVig | HDIVig-refractory and severe disease | Lenalidomide + dexamethasone + HDIVig | 3 mo§ | PR | CR (IF negative) |
| 7 | 28 | F | PE, dilated cardiomyopathy, and impaired consciousness | λ, 5.3 g/L; MGUS | HDIVig | HDIVig-refractory and severe disease | Lenalidomide + dexamethasone + HDIVig | 3 mo§ | PR | CR (IF negative) |
| Patient no. | Age, y | Sex | Characteristics | MG, level; hemopathy | Previous treatment | Status | Treatment | Duration/protocol | Clinical response | Hematologic response |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 65 | F | PE, myalgia, and arthralgia | κ, 4 g/L; MGUS | HDIVig | HDIVig-refractory disease | Bortezomib + cyclophosphamide + dexamethasone and HDIVig | 4 mo* | PR | CR (IF negative) |
| 2 | 61 | F | PE, CTS, and arthralgia | λ, 4 g/L; MGUS | HDIVig | HDIVig- refractory disease | Bortezomib + melphalan + dexamethasone | 1 mo† | NR, death | NR |
| 3 | 50 | M | PE, recurrent DNS, and CTS | κ, 3.9 g/L; MGUS | HDIVig | HDIVig-refractory and severe disease | Bortezomib + cyclophosphamide + dexamethasone | 2 mo* | NR | PR |
| Bortezomib + lenalidomide + dexamethasone | 5 mo‡ | PR (almost complete) | CR (IF negative) | |||||||
| 4 | 43 | M | PE, DNS, and CTS | λ, 4.5 g/L; smoldering myeloma | HDIVig + CT | HDIVig-refractory disease | Lenalidomide + dexamethasone + HDIVig | 6 mo§ | PR (almost complete) | CR (IF negative) |
| 5 | 63 | F | PE, DNS, epilepsy, and ischemic cardiopathy | κ, 11 g/L; MGUS | — | Severe disease | Bortezomib + dexamethasone | 4 mo¶ | CR | CR (IF negative) |
| 6 | 29 | F | PE, DNS, livedo, and GAL | λ, 7 g/L; MGUS | HDIVig | HDIVig-refractory and severe disease | Lenalidomide + dexamethasone + HDIVig | 3 mo§ | PR | CR (IF negative) |
| 7 | 28 | F | PE, dilated cardiomyopathy, and impaired consciousness | λ, 5.3 g/L; MGUS | HDIVig | HDIVig-refractory and severe disease | Lenalidomide + dexamethasone + HDIVig | 3 mo§ | PR | CR (IF negative) |
CT, corticosteroids PO; CTS, carpal tunnel syndrome; IF, immunofixation; IVP, intravenous pyelogram; livedo, livedo racemosa; PE, papular eruption.
Bortezomib, 1.3 mg/m2 IVP on days 1, 4, 8, and 11 + cyclophosphamide, 300 mg/m2 per day PO on days 1, 8, 15, and 22 + dexamethasone 40 mg PO on days 1, 8, 15, and 22.
Melphalan, 10 mg/m2 PO on days 1 through 4 + bortezomib, 1.3 mg/m2 IVP on days 8, 15, and 22 + oral dexamethasone, 40 mg/d on days 1 through 4.
Bortezomib, 1.3 mg/m2 IVP on days 1, 4, 8, and 11 + lenalidomide, 25 mg/d on days 1 through 14 + dexamethasone, 20 mg/d on days 1, 2, 4, 5, 8, 9, 11, and 12.
Lenalidomide, 25 mg/d on days 1 through 21 + dexamethasone, PO 40 mg on days 1, 8, 15, and 22 + HDIVig, 2 g/kg per month.
Bortezomib, 1.3 mg/m2 IVP on days 1, 4, 8, and 11 + dexamethasone, 40 mg on days 1, 2, 3, 4, 8, 9, 10, and 11.
DNS
The specific management of DNS (n = 6) is summarized in Table 2. Hospitalization in the intensive care unit was required for all 6 patients because of coma and/or generalized epilepsy. All patients received a combination of antiepileptic drugs. The most common specific therapy was plasmapheresis with or without HDIVig and steroids, which induced a complete and rapid resolution of DNS symptoms (4/5). After the resolution of DNS, 2 patients received maintenance therapy with HDIVig, lenalidomide, and dexamethasone, 1 patient received maintenance therapy with bortezomib, dexamethasone, and HDIVig, and 1 patient received maintenance therapy with HDIVig, bortezomib, lenalidomide, and dexamethasone with a long-term skin response, no neurological recurrence, and a median time without relapse of 1.5 years (range, 1-2 years). One patient died of severe sepsis and specific heart involvement following chemotherapy treatment (bortezomib, melphalan, and dexamethasone).
Prognosis
At 3 years, the overall survival of all 33 patients was 97%, and overall survival without DNS or specific mucinous cardiomyopathy was 82% (Figure 2).
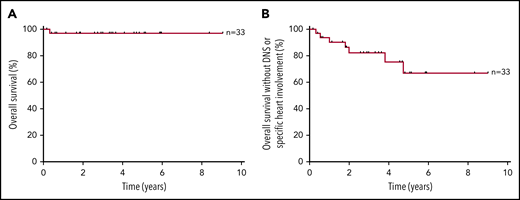
Kaplan-Meier analysis of survival. (A) Overall survival. (B) Overall survival without DNS or specific heart involvement.
Kaplan-Meier analysis of survival. (A) Overall survival. (B) Overall survival without DNS or specific heart involvement.
Discussion
In this largest series of scleromyxedema, we reported an overall improved survival rate compared with previous studies4,5 and the efficacy of HDIVig therapy in nonsevere patients. We also showed the efficacy and safety of a plasma cell–directed regimen in severe, refractory, or relapsed scleromyxedema.
Our series of patients highlights the high frequency of central nervous system injuries in scleromyxedema, especially DNS. DNS usually starts as a “flu-like” syndrome and fever, followed by multiple neurological symptoms, such as convulsions, seizures, and coma.12-15 Other central neurological symptoms have been described without typical DNS features (isolated epilepsy and acute psychosis). The mechanisms that cause this syndrome are not understood. Usually, cerebrospinal fluid analysis and brain imaging are normal, as in our series. Brain mucin deposition has never been found in brain biopsies or postmortem autopsies. An acute demyelinating process could be involved, as shown in sensitive brain imaging, such as cerebral MRI or autopsies.12,15,16 Increased IL-6 levels were noted in the cerebrospinal fluid in 2 patients.17,18 Various skin features have been described in scleromyxedema, such as mostly papular eruptions that are localized at the glabella and auricular zone and sclerodermatous changes. Cutis laxa–like eruptions are another frequent skin feature that could be related to the fragmentation of elastic fibers.19-21 In our study, associated hematological malignancies were present in 12% of patients, which was consistent with previous publications.4,5 One patient had refractory cytopenia with multilineage dysplasia, which might have been an incidental finding that was unrelated to MG.
For the past 30 years, the mortality of scleromyxedema patients has gradually decreased from 35% in 1995,5 to 23.8% in 2013,4 and to 3% in our current study, despite similar frequencies of DNS and underlying hematologic malignancies. This observation could have been partially influenced by the progress of patient management in intensive care units; however, the main contributing factor could be the increase in the use of HDIVig therapies. HDIVig-induced clinical responses are not usually associated with a hematological response because HDIVig has no effect on serum MG levels. Therefore, maintenance therapy is often needed, given the transient efficacy of HDIVig. Considering the small number of patients in the immunomodulatory drug group, a formal comparison of patients treated with HDIVig and immunomodulatory drugs was not performed.
Although HDIVig has been reported to have high efficacy and a good safety profile, and it has now been validated in skin-limited or nonsevere scleromyxedema, there is no evidence regarding the prevention of the severe forms of this disease, such as DNS, and the risk of progression to multiple myeloma. Targeting the underlying plasma cell hemopathy seems to be an interesting strategy and could be important for altering the disease outcome.
Scleromyxedema is a type of MG of clinical significance, which is a recently described entity.2,3,21 Similar to patients with type I cryoglobulinemia,22 amyloid light-chain amyloidosis,23 or MG-associated xanthoma,24 scleromyxedema patients who are refractory to HDIVig treatment and who have severe systemic forms of the disorder, such as DNS or mucinous cardiomyopathy, may require plasma cell–directed therapies. Our data highlight the benefit of lenalidomide or bortezomib + dexamethasone combined with maintenance infusions of HDIVig to induce sustained hematological and clinical responses with good control of severe injuries and better safety than the old protocols that used alkylating agents. Further follow-up with more treated patients is needed to determine whether the withdrawal of HDIVig is possible in patients who achieve a hematological CR after antiplasma cell therapy. All of these results support the use of such antiplasma cell drugs in severe, refractory, or relapsed scleromyxedema. When these emerging strategies fail, autologous stem cell transplantation has shown good efficacy as salvage therapy in a few patients with relatively good safety.25-29 New specific therapies that target plasmocyte antigens, such as CD38, could support their use in severe and refractory scleromyxedema in the near future.30 Because of the small number of patients treated with each therapeutic regimen, caution is needed when drawing conclusions about the definitive efficacy of each drug used in this study.
Based on a systematic analysis of the literature and the analysis of our patient data, we can now provide a therapeutic management algorithm for scleromyxedema (Figure 3). In nonsevere scleromyxedema (skin limited or no complicated extracutaneous disease), HDIVig (2 g/kg) every month until the best clinical response is achieved,4,6,7 followed by maintenance for ≥6 months, appears to be the most effective treatment. HDIVig-refractory or severe forms of this disease may require plasma cell–directed drugs, such as bortezomib + dexamethasone31-34 or lenalidomide + dexamethasone.35,36 In cases of DNS, hospitalization in the intensive care unit in a reference center is necessary, with treatment consisting of a combination of HDIVig, high-dose steroids, and plasmapheresis.14,15,37,38 Rare cases of the spontaneous regression of DNS have been described.15 Maintenance therapy with HDIVig and a plasma cell–directed regimen should be initiated to avoid disease recurrence. In the case of plasma cell–directed drug use, the response should be analyzed at the clinical and hematological levels. Hematological responses are assessed according to the International Myeloma Working Group consensus criteria.39

Proposed algorithm for the therapeutic ± management of MG-associated scleromyxedema. *Bortezomib- or lenalidomide-based regimen. †Response will be assessed clinically and hematologically according to International Myeloma Working Group criteria.39
Proposed algorithm for the therapeutic ± management of MG-associated scleromyxedema. *Bortezomib- or lenalidomide-based regimen. †Response will be assessed clinically and hematologically according to International Myeloma Working Group criteria.39
Generally, plasma cell–directed strategies, which target the underlying hemopathies, in MG-related scleromyxedema could be interesting to evaluate.
We found a significant increase in TGF-β and collagen-1a in the skin samples of patients with scleromyxedema. A previous immunochemistry study showed an increase in procollagen 1 in the skin of patients with scleromyxedema compared with patients with systemic nephrogenic fibrosis.40 As shown in mouse models of systemic sclerosis, the great efficacy of HDIVig in skin fibrosis could be linked to a local reversion of these 2 factors in the dermal tissue.41 A recent study highlighted the role of Tc17 lymphocytes and their correlation with skin involvement and confirmed the local decrease in TGF-β after HDIVig treatment.42 Prospective studies targeting fibrotic pathways or plasma cell clones are highly warranted for scleromyxedema. The following experiments could be conducted to gain additional insights into the physiopathology of scleromyxedema and to guide therapeutic strategies: (1) analyze the secretome serum (mass spectrometry) and purify the MG protein to determine whether soluble factors or MG are responsible for fibroblast proliferation or mucin production, (2) perform B-cell receptor repertoire analysis with flow cytometry–sorted clonal plasmocytes, (3) analyze the conformational specificity of monoclonal Ig using crystallography, (4) identify specific plasmocyte mutations using next-generation sequencing, and (5) perform in-depth analysis of skin-specific fibroblast modifications using RNA sequencing.
In summary, this study provides new insights into paraprotein-targeting strategies in patients with scleromyxedema, particularly in patients with severe, refractory, and relapsed disease. Prospective studies targeting plasma cell clones and/or fibrotic pathways are warranted for scleromyxedema management.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: T.M., B.A., J.-D.B., A.M., and O.F. designed and performed the research, analyzed data, and wrote the manuscript; T.M., B.A., J.-D.B., C.B.L., A.S., B.C., B.S., M.J., L.M., P.A., D.L., C.F., E.S., M.R., V.D., M.D., P.H., M.B.-B., T.P., C.d.M., R.Y.T., O.H., A.D., S.B., S.D., O.C., F.B., J.-L.S., D.T.-B., P.M., C.Z., F.L., E.B., N.L., F.L.B., M.L.M., M.T., A.T., R.P., S.P., A.D.M., M. Battistella, A.M., and O.F. recruited patients; T.M., B.A., J.-D.B., A.M., and O.F. analyzed data and reviewed the manuscript; A.O. and M. Bagot performed histopathologic studies, analyzed data, and reviewed the manuscript; T.M., S.P., and L.M. performed PCR on skin biopsy and analyzed data; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the Etude des Maladies Systémiques en Dermatologie appears in “Appendix.”
Correspondence: Thibault Mahévas, Service de Médecine Interne, Hôpital Saint Antoine, AP-HP, 184 Rue du Faubourg Saint-Antoine, 75012 Paris, France; e-mail: thibault.mahevas@gmail.com.
Appendix
Members of the Etude des Maladies Systémiques en Dermatologie: Didier Bessis, Nadège Cordel, Dan Lipsker, Jean-David Bouaziz,Charles Cassius, and François Chasset.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal