


Abstract
The TEMPI syndrome is a rare and acquired disorder characterized by 5 salient features, which compose its name: (1) telangiectasias; (2) elevated erythropoietin and erythrocytosis; (3) monoclonal gammopathy; (4) perinephric fluid collections; and (5) intrapulmonary shunting. Complete resolution of symptoms following treatment with plasma cell-directed therapy supports the hypothesis that the monoclonal antibody is causal and pathogenic. Understanding the basis of the TEMPI syndrome will depend on the identification of additional patients and a coordinated international effort.
History
The telangiectasias, elevated erythropoietin and erythrocytosis, monoclonal gammopathy, perinephric fluid collections, and intrapulmonary shunting (TEMPI) syndrome was born out of serendipity and international collaboration. In 2008, we met a man with an undiagnosed medical condition who had traveled to Boston to seek another medical opinion. After an extensive workup, this case, “A 49-year-old man with erythrocytosis, perinephric fluid collections, and renal failure,” was published in the Case Records of the Massachusetts General Hospital.1 By special request, the editor of the Case Records at the time, Nancy Lee Harris, allowed us to conclude the manuscript with the statement “We request that any reader with thoughts about the diagnosis, further evaluation, or treatment contact David Sykes.” Within weeks of publication, 2 more patients were identified, ultimately resulting in the publication of the index paper on the TEMPI syndrome with 3 living patients and an additional 3 patients gleaned from literature case reports.2
As additional patients became identified, the association of erythrocytosis and plasma cell dyscrasias was discussed3 and the TEMPI syndrome was recognized as a monoclonal gammopathy of cutaneous significance4 as well as a monoclonal gammopathy of clinical significance.5 Ultimately, the TEMPI syndrome has taken a place alongside the polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy or M proteins, and skin abnormalities syndrome as “plasma cell neoplasms with associated paraneoplastic syndrome” in the World Health Organization Classification of Tumors of Hematopoietic and Lymphoid Tissues, 4th edition. On November 30, 2018, the first international TEMPI syndrome symposium was held in San Diego, CA.
The syndrome
A total of 22 patients with the TEMPI syndrome have been identified (as of December 2019), many of whom have been described in individual case reports6-14 spanning 11 countries across the globe (supplemental Figure 1, available on the Blood Web site). The TEMPI syndrome is an acquired disorder that generally manifests in the fourth or fifth decade of life. The syndrome occurs in both men and women without any discernable ethnic or geographic predisposition.
Almost invariably, these patients first present with erythrocytosis and telangiectasias and many have been erroneously diagnosed with polycythemia vera and initiated on programs of therapeutic phlebotomy. In all patients, laboratory values are notable for an elevated serum erythropoietin and the lack of a JAK2 mutation. In those patients who have been tested, hemoglobin electrophoresis and hemoglobin oxygen affinity testing have been normal, and no underlying genetic abnormalities have been identified.
Telangiectasias are seen most prominently on the face and upper back and chest (Figure 1). The hands are also commonly affected, whereas the lower extremities seem to be spared of telangiectasias.
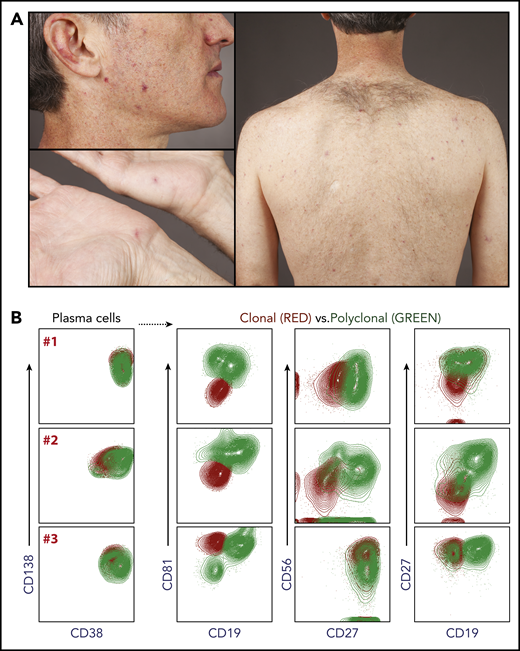
Telangiectasias and flow cytometry. (A) Telangiectasias are most prominent on the face and the trunk and rarely seen on the lower extremities. (B) Extended phenotyping of bone marrow plasma cells from 3 patients (designated A, B, C) with TEMPI syndrome has been performed by flow cytometry using a panel of markers including: CD38, CD138, CD19, CD27, CD56, and CD81. The loss of CD19 appears to be a consistent feature of the monoclonal plasma cell population.
Telangiectasias and flow cytometry. (A) Telangiectasias are most prominent on the face and the trunk and rarely seen on the lower extremities. (B) Extended phenotyping of bone marrow plasma cells from 3 patients (designated A, B, C) with TEMPI syndrome has been performed by flow cytometry using a panel of markers including: CD38, CD138, CD19, CD27, CD56, and CD81. The loss of CD19 appears to be a consistent feature of the monoclonal plasma cell population.
The hallmark feature of the TEMPI syndrome is a monoclonal gammopathy. There does not appear to be any immunoglobulin or light chain specificity (unlike the polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy or M proteins, and skin abnormalities syndrome in which the monoclonal antibody is almost always λ-restricted15 ). Despite the monoclonal gammopathy, the patients do not exhibit any of the calcium, renal failure, anemia, and bone lesion criteria diagnostic for multiple myeloma and do not have more than 10% plasma cells in the bone marrow at presentation.3 The serum free light chains and serum free light chain ratio have been generally normal or close to normal.
Over many years, the erythropoietin level tends to progressively increase, preceding the development of the features of intrapulmonary shunting and perinephric fluid collections. The serum erythropoietin measurements can be extremely high (>5000 mIU/mL; normal range, 3-19 mIU/mL), driving a predictable secondary erythrocytosis. Most TEMPI syndrome patients have been initiated empirically on programs of therapeutic phlebotomy leading to iron deficiency and microcytosis.16
The pulmonary shunting has been termed “microscopic” in that high-resolution computed tomography scans of the chest are normal and echocardiography using agitated saline contrast shows the late appearance of left-sided bubbles. The pulmonary shunting is best quantified by 99mTc macro-aggregated albumin scintigraphy. The pulmonary shunting is accompanied by a decrease in the resting oxygen saturation and ultimately a dependence on supplementary oxygen. It is typically for reasons of progressive hypoxia that patients have required the initiation of treatment.
The etiology of the perinephric fluid collections remains unclear. Their sonographic characteristics have been reviewed and compared with other fluid accumulations.17 When sampled, the fluid is sterile with no cells and no protein. It does not appear to be lymphatic fluid or chyle, and the fluid has the same chemical (eg, sodium, chloride, potassium) composition as serum.1 Early in the identification of the TEMPI syndrome, 2 patients with large perinephric fluid collections underwent surgical fenestration to drain the accumulating fluid into the abdominal cavity.
A subset of patients with TEMPI syndrome have developed venous thromboses as has also been described in patients with monoclonal gammopathy of undetermined significance and multiple myeloma18 ; arterial thromboses have not been seen to date. Although the telangiectasias appear to be confined to the skin, 1 patient did develop extensive bleeding from gastrointestinal arteriovenous malformations late in the disease and 2 patients have suffered spontaneous intracranial hemorrhages.
Based on the patient characteristics to date, we have proposed diagnostic criteria as outlined in Table 1.
TEMPI syndrome diagnostic criteria
| Criteria |
|---|
| Major |
| Telangiectasias |
| Elevated erythropoietin and erythrocytosis |
| Monoclonal gammopathy |
| Minor |
| Perinephric fluid |
| Intrapulmonary shunting |
| Other |
| Venous thrombosis |
| Criteria |
|---|
| Major |
| Telangiectasias |
| Elevated erythropoietin and erythrocytosis |
| Monoclonal gammopathy |
| Minor |
| Perinephric fluid |
| Intrapulmonary shunting |
| Other |
| Venous thrombosis |
Pathology
In 2 patients, the telangiectatic lesions have been biopsied without unusual features. The peripheral blood smear is morphologically unremarkable. The bone marrow biopsy typically shows findings consistent with those of monoclonal gammopathy of undetermined significance/smoldering myeloma, with only a mild increase in plasma cells (5% to 10% by immunohistochemistry).3 Given the low percentage of plasma cells, flow cytometry is often the best method to establish evidence of plasma cell clonality. In the patients for which this has been done with an extended panel of antibodies,19 the clonal plasma cells have been easily distinguished based on their pattern of cell surface staining (Figure 1).
In patients undergoing bone marrow biopsy for suspected polycythemia vera, the absence of abnormalities associated with myeloproliferative neoplasms (eg, atypical megakaryocytic hyperplasia, platelet abnormalities, hypercellularity) is remarkable, and in at least 1 case triggered the workup for TEMPI as the explanation for the patient’s erythrocytosis.
Treatment
Treatment of the first patient with TEMPI syndrome was done empirically and out of necessity in the setting of worsening hypoxia resulting from life-threatening pulmonary shunting. After much discussion, it was believed to be extraordinarily unlikely that each of the 6 initial patients would have an innocent bystander monoclonal gammopathy.2 For this reason, plasma cell–directed therapy in the form of bortezomib was used, with both complete and partial responses reported.11,20-22 Autologous transplantation following a melphalan-based conditioned regimen was also successful in 1 patient,23 though not in another who subsequently went on to achieve a complete response following treatment with daratumumab.24 Most recently, successful treatment with lenalidomide was reported.14
Treatment responses have been rapid and remarkable. Patients who have responded to treatment with eradication of their monoclonal gammopathy have also seen complete resolution of all of the other symptoms of the TEMPI syndrome. Three patients with severe intrapulmonary shunting, requiring continuous supplemental oxygen, have had complete reversal of their shunt within months of initiating effective therapy. The telangiectasias and intrapulmonary shunting seem to respond within the same time frame, suggesting that these might be part of the same abnormal angiogenic process.
In those patients who have relapsed after initial therapy, the most sensitive marker of relapse appears to be the increase of the serum erythropoietin, followed shortly thereafter by detection of the monoclonal gammopathy and then reappearance of skin telangiectasias.
Understanding the limitations of proposing treatment guidelines in such a rare condition, we would recommend initial treatment with daratumumab (where available) or with bortezomib, both of which have been effective and well-tolerated. As the number of patients and therapeutic experience continues to grow, we would of course welcome discussions around individual cases.
Pathophysiology
As described, the recognition that the TEMPI syndrome is acquired and completely reversible with effective plasma cell–directed therapy has implicated either the monoclonal antibody or the monoclonal plasma cell in the pathogenesis of the disease. We initially wondered whether prolonged exposure to extraordinarily high levels of serum erythropoietin might play a role, but the other TEMPI symptoms have not been reported in animals or humans exposed to supraphysiologic erythropoietin.25
What about the possibility of a plasma cell–secreted circulating factor? This is also an attractive hypothesis, although there is presently no known single chemokine or cytokine that could explain the constellation of symptoms of the TEMPI syndrome.
The monoclonal gammopathies of undetermined significance were recently reviewed in Blood.26 The concept of a monoclonal gammopathy of clinical significance, postulated more than a decade ago in Blood27 and recently updated,5 mirrors our current hypothesis. This hypothesis would suggest that the TEMPI syndrome is effectively an autoimmune disease in which the monoclonal antibody activates or inactivates an as-yet-unknown pathway, leading to the unregulated production of erythropoietin and the development of telangiectasias, pulmonary shunting, and perinephric fluid collections.
It is intriguing to postulate that the monoclonal gammopathy may be perturbing the oxygen-sensing machinery and in some way contributing to the stabilization of HIF1-α, ultimately driving the dramatic expression of erythropoietin.28-30
How does this hypothesis fit within our current understanding of other antibody-mediated autoimmune diseases? In the case of more common hematologic diseases such as immune thrombocytopenia purpura, autoimmune hemolytic anemia, and antiphospholipid antibody syndrome, the antibody targets a specific epitope with high affinity and the level of the antibody remains so low as to be undetectable on serum protein electrophoresis and immunofixation.
The observation that the monoclonal antibody in TEMPI syndrome is detectable via conventional electrophoresis and therefore present at a concentration at least 100 times higher than the antibody in other autoimmune disorders argues that it is either not involved in the pathogenesis of the disease or does not have a high-affinity protein target. We postulate that the antibody may (1) be low affinity, (2) have a nonprotein target, or (3) have an intracellular target.
The antigenic target of autoantibodies has been identified in cases of seminoma-associated paraneoplastic encephalitis31 and autoimmune pancreatitis.32 Interestingly, a lipid-based autoantigen has also been implicated in driving the proliferation of plasma cells in rare circumstances.33
In the case of TEMPI syndrome, our efforts have focused on searching for other circulating cytokines or on trying to identify a peptide or protein target of the monoclonal gammopathy. Techniques have including1 immunofluorescence of fresh-frozen normal tissue samples,2 immunohistochemistry of fixed normal tissue samples,3 immunoblotting of denatured tissue lysates,4 and peptide and protein microarrays, although none of these have identified a convincing antigenic target.
Gene expression analysis (RNA sequencing) and mutational analysis (whole exome sequencing) of bulk-sorted plasma cell populations in 3 patients has been unrevealing. We have since turned to techniques of single-cell plasma cell sequencing and of cloning the heavy and light chain regions of the monoclonal antibody. We are hopeful that a panel of monoclonal antibodies generated from TEMPI patients will increase the likelihood of antigen identification.
Conclusion
The TEMPI syndrome should be considered in a patient with longstanding and unexplained erythrocytosis with the combination of an elevated erythropoietin and monoclonal gammopathy, prompting a careful examination for telangiectasias as well as imaging to evaluate for perinephric fluid collections.
Just as the description of the TEMPI syndrome was born out of international collaboration, so will an understanding of its pathophysiology. Ultrarare diseases such as the TEMPI syndrome highlight the need for international registries and international institutional review boards to facilitate the collection and storage of research samples across the globe.
The online version of this article contains a data supplement.
Acknowledgments
The authors specifically thank their patients with whom they have worked together to learn about the TEMPI syndrome, and the physicians and care teams across the world who have helped to identify and to treat this very rare group of individuals. The authors also give special thanks to Flavia Rosado at University of Texas Southwestern, and to Jens Wrammert at Emory University who have been closely involved with the research efforts, and to Olga Kharchenko whose artistry was behind the visual abstract.
Authorship
Contribution: D.B.S., C.O., and W.S. conceived, wrote, edited, and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David B. Sykes, Massachusetts General Hospital, 185 Cambridge St, CPZN 4238, Boston, MA 02114; e-mail: dbsykes@partners.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal